

Comportamiento del SDS localizado en la región interfacial del sistema agua/n-octano. Un estudio usando dinámica molecular
Behaviour of the SDS located at the interfacial region of the water/n-octane system. A study using molecular dynamics
ACI Avances en Ciencias e Ingenierías
Universidad San Francisco de Quito, Ecuador
ISSN: 1390-5384
ISSN-e: 2528-7788
Periodicidad: Bianual
vol. especial, núm. 1, 2020
Recepción: 02 Marzo 2016
Aprobación: 08 Diciembre 2016
Resumen: En este trabajo, usando dinámica molecular se determinaron las propiedades interfaciales y el comportamiento del Dodecil Sulfato de Sodio (SDS) ubicado en la región interfacial de los sistemas vacío/agua y agua/n-octano. La tensión interfacial fue estimada con el modelo propuesto por Kirkwood y Buff [23]. A su vez, los espesores de película interfacial fueron determinados usando los criterios 10-90 y 90-90. Además, el área por molécula fue estimado con la variación de la presión superficial en función de la concentración del surfactante. En los sistemas vacío/SDS/agua, el área 2 por molécula del SDS fue obtenida con dos procedimientos diferentes. Los valores fueron 53.3 A y 54.3 A , respectivamente. Para los sistemas agua/n-octano y agua/SDS/n-octano, los espesores de película interfacial aumentan en función del número de moléculas de surfactantes presentes en la región interfacial. Los resultados obtenidos son consistentes con datos medidos por experimentación. ' ISSN (Impreso): 1390-5384“ ISSN (Electrónico): 2528-7788
Palabras clave: Dinámica molecular, espesor de película interfacial, tensión interfacial.
Abstract: In this paper, molecular dynamics simulations was used to determine the interfacial properties and behavior of Sodium Dodecyl Sulfate (SDS) located at the interfacial region of the vacuum/water and water/n-octane systems. The interfacial tension was estimated with the model proposed by Kirkwood and Buff [23]. In turn, to determine the interfacial film thickness were used the criteria 10-90 and 90-90. Also, the area per molecule was calculated with the variation of the surface pressure in function of concentration of the surfactant. Moreover, the values of area per molecule of SDS in water estimated with two procedure differents. The obtained values were 53.3 A and 54.3 A , respectively . Finally, for water/n-octane and water/sds/n-octane systems the thickness of the interfacial film increase in function of the number of molecules of surfactants present in the interfacial region. The results are consistent with experimental values.
Keywords: Molecular Dynamic, interfacial film thickness, interfacial tension.
Introducción
Los surfactantes, son moléculas anfifílicas que tienen una cabeza polar de naturaleza hidrofílica y una cadena hidrocarbonada tipo lipofílica [1,2]. Estas moléculas, tienen la capacidad de reducir la tensión interfacial de las interfaces vapor/líquido, líquido/líquido y líquido/sólido.
La adsorción de surfactantes en la región interfacial depende de sus propiedades anfifílicas únicas, las cuales son conocidas como el balance entre las fuerzas hidrofílicas y lipofílicas de la cabeza polar y la cadena lipofílica. El comportamiento de los surfactantes ubicados en las regiones interfaciales juegan un papel importante en muchas aplicaciones como detergencia, flotación mineral, dispersión de sólidos, recuperación de crudos, dispersión de nanopartículas, entre otros. Todas estas aplicaciones han motivado diversos estudios para describir agregados y monocapas de surfactantes en diferentes interfaces [3].
A nivel experimental, el dodecil sulfato de sodio (SDS, C12H25OSO3-Na+) es uno de los surfactantes iónicos más utilizados en los estudios de agregación micelar, estabilidad de espumas y formación de monocapas. A su vez, el SDS tiene alto valor de solubilidad en agua lo cual expresa su alto nivel hidrofílico [4]. Experimentalmente, se ha estudiado la adsorción de SDS en las regiones interfaciales aire/aceite, aire/agua [5-7] y agua/grafito usando espectroscopía de frecuencia vibracional, reflac- tancia de neutrones y microscopía de fuerza atómica con la finalidad de estimar la orientación molecular de los surfactantes en la región interfacial [8,9]. Además, el SDS tiene la capacidad de reducir la tensión interfacial de sistemas vapor/agua desde 72 hasta 39 mN/m para altas concentraciones en agua y en sistemas agua/aceite como agua/hexadecano desde 52 hasta 10 mN/m [5,6]. También, se han reportado su área mánima ocupada por molécula de 52.0 y 50.3 Á en las regiones interfaciales aire/agua y agua/n-octano, respectivamente [2,7].
Con el desarrollo computacional, las técnicas de simulación como dinámica molecular ha sido muy utilizadas para estudiar el auto ensamblaje de surfactantes en las regiones interfaciales aire/agua y aire/aceite a nivel atomístico [10-14]. En estos trabajos, se determinaron las tensiones interfaciales, las presiones superficiales y algunas propiedades estructurales. Sin embargo, existen pocos trabajos donde se haya determinado la energía necesaria para la formación de la interfaz del sistema vacío/SDS/agua [15] . Adicionalmente, las propiedades estructurales y el mecanismo de la agregación del SDS en agua han sido estudiadas con dinámica molecular [16,17].
Por tal motivo, en este trabajo, se determinaron las propiedades interfaciales (presión superficial, película interfacial y área por molécula) de los sistemas vacío/SDS/agua y agua/SDS/n-octano, con la finalidad de profundizar en el comportamiento del surfactante SDS desde el punto de vista molecular.
Métodos
Modelos de energía potencial
En simulaciones con dinámica molecular, los sistemas moleculares son descritos por un modelo de energía potencial denominado force field [18]. El modelo de energía potencial incluye los términos enlazantes y no enlazantes, los cuales incluyen las fuerzas electrostáticas y de van der Waals [18]. Por lo tanto, la energía de interacción total entre moléculas puede ser escrita como:
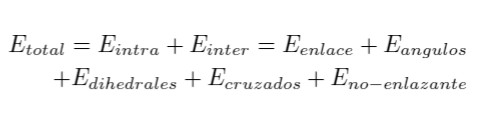 (1)
(1)Donde,, y son las energías intramolecular, intermolecular y total, respectivamente.
En este trabajo, las moléculas de SDS y n-octano fueron descritas usando el modelo de energía potencial GRO- MOS53A6 [18]. Los parámetros de este modelo de energía potencial fueron generados con el software Automated force field Topology Builder (ATB) [19]. En el force field GROMOS53A6, los términos asociados a las interacciones no enlazantes fueron calculados mediante la ecuación 2.
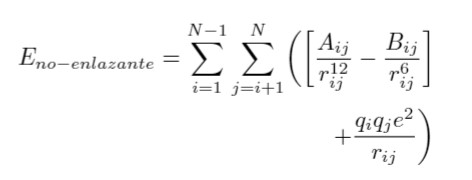 (2)
(2)Donde, ,,, and son los parámetros de energía, cargas atómicas y distancia entre partículas i y j , respectivamente.
Para el agua, el modelo de punto de carga simple (Simple Point Charge, SPC) fue usado en nuestra simulación (SPC model)[20]. En este modelo, la distancia OH es 0.100 nm y el ángulo HOH es 109.47o. Además, existe una ínica interacción de Lennard-Jones entre los sitios del oxígeno. Los parámetros de Lennard-Jones y cargas atómicas en el modelo SPC son σ(O) = 0,317 nm,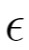 (O) = 0,650 kJ/mol, q(H) = 0,4100e y q(O) =- 0.8200e. El modelo SPC permite predecir resultados exactos de las propiedades del agua como la densidad y la difusión en fase líquida.
(O) = 0,650 kJ/mol, q(H) = 0,4100e y q(O) =- 0.8200e. El modelo SPC permite predecir resultados exactos de las propiedades del agua como la densidad y la difusión en fase líquida.
Perfiles de densidad y película interfacial
El perfil de densidad de un sistema interfacial describe la no homogeneidad de una interfaz y permite localizar la región interfacial y el espesor de la misma. Para una interfaz líquido/líquido, la región interfacial comienza, donde la densidad de ambos líquidos disminuye con respecto al seno del líquido puro. Generalmente, los perfiles de densidad de una partícula tipo i, a lo largo de la dirección perpendicular a la interfaz se describe por la ecuación 3.
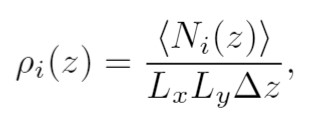 (3)
(3)Donde, es el perfil de densidad a lo largo del eje Z, 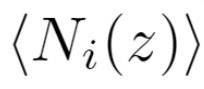 es el número de partículas tipo i entre y en el tiempo t [21]. En este trabajo, fue escogido a un valor igual a 0.01 nm.
es el número de partículas tipo i entre y en el tiempo t [21]. En este trabajo, fue escogido a un valor igual a 0.01 nm.
Tensión interfacial
La propiedad macroscópica más importante para definir un sistema interfacial es la tensión superficial. Los métodos usados para calcular la tensión superficial son basados sobre la definición mecánica [22-26]. La primera forma explícita desarrollada por Kirkwood and Buff expresa a los tensores de presión como una función derivada de la energía potencial [23]. En este caso, la tensión superficial es definida por la ecuación 4.
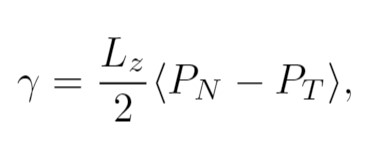 (4)
(4)Donde, PN y PT son los componentes normal y tangencial de la presión, respectivamente. Lz es la dimensión de la celda de simulación a lo largo el eje Z. El componente normal PN es igual a Pzz, mientras el componente tangencial PT es dado por 1/2(Pxx + Pyy). Los tensores de presión Pxx, Pyy y Pzz, desde el punto de vista molecular, son definidos de forma general mediante la ecuación 5.
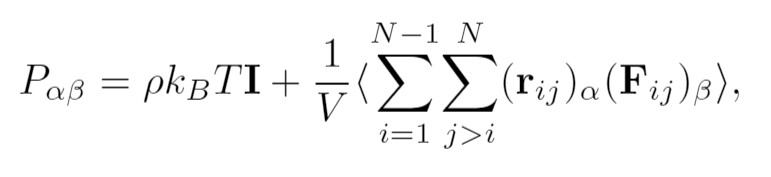 (5)
(5)Donde I es el tensor unitario, kB es la constante de Boltzmann, T es la temperatura, y es la densidad en número. Los términos a y ^ representan las direcciones X, Y, y Z. En esta ecuación 5, rij es el vector entre el centro de masa de la molécula i y j. El término Fij es la fuerza intermolecular entre moléculas i y j, el cual se expresa como la suma de todas las fuerzas interactuantes entre estas moléculas [27].
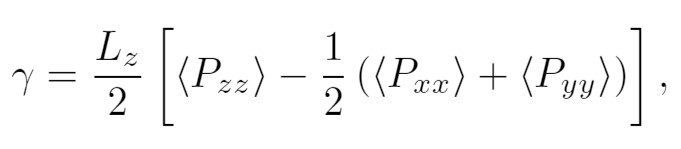 (6)
(6)Generalmente, en dinámica molecular la tensión interfacial se calcula usando el tensor presión promedio. En este caso, la tensión interfacial es determinada a lo largo del eje Z mediante la ecuación 6.
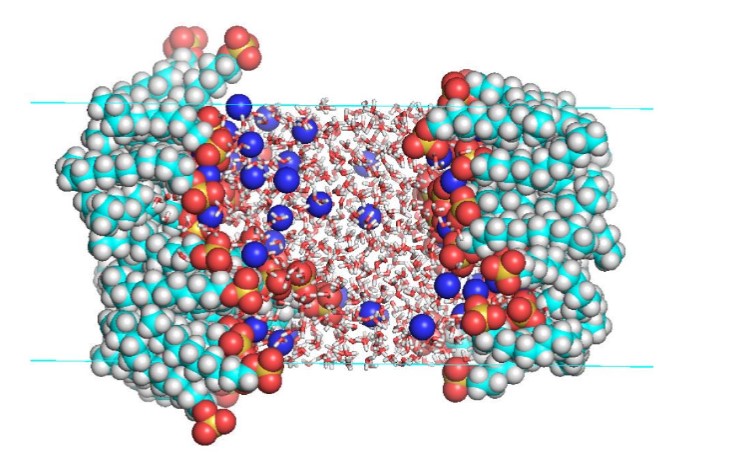
Sistema SDS/agua/SDS previamente relajados utilizando el programa Gromacs-4.5.5.
Sistemas simulados
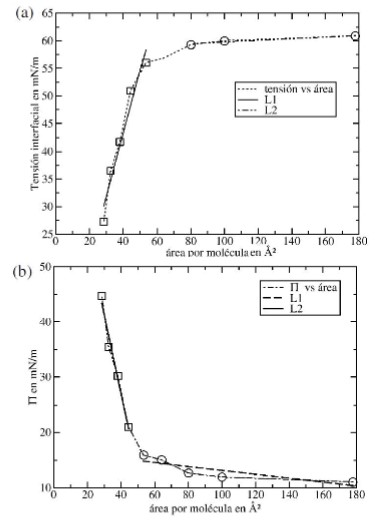
En este estudio se utilizaron dos tipos de sistemas. Para los sistemas vacío/SDS/agua se construyeron celdas periódicas rectangulares de longitudes Lx = Ly = 3 nm y Lz = 30 nm, con una capa de agua constituida por 1000 moléculas ubicada en el centro de la celda a lo largo del eje z. Las monocapas de surfactantes contenían 9, 12, 16, 20, 25, 30 moléculas de SDS y se colocaron a los extremos de la capa de agua como se puede observar en la figura 1.
Las dimensiones de las celdas periódicas de agua y noctano fueron de 5x5x4 nm3. Estas celdas periódicas se unieron formando una caja rectangular de 5x5x8 nm3. El número de moléculas de SDS presentes en las monocapas fueron 9, 12, 16, 20, 25 y 36. Adicionalmente, las moléculas de SDS y n-octano han sido descritas con el modelo de energía potencial GROMOS-53A6 [28,29]. Para el caso del SDS, los parámetros usados corresponden al modelo de Berkowitz y colaboradores [30,31]. En cambio, el modelo SPC fue usado para simular las moléculas de agua. Al final de las simulaciones, los sistemas estaban confinados en cajas rectangulares de 4x4x8.2 nm3. Seguidamente, los sistemas agua/n-octano y agua/SDS/n-octano fueron construidos usando 188 moléculas de n-octano y 1640 moléculas de agua. En la figura 2, se muestra un ejemplo de los modelos construídos. La construcción de los sistemas simulados se realizó usando las herramientas genbox, genconf y editconf del programa Groningen Machine for Chemical Simulations (GROMACS) versión 4.5.5 [32-35].
Condiciones de las simulaciones.
Todas las simulaciones se realizaron con el programa Gromacs 4.5.5. Los sistemas fueron periódicos en las coordenadas XYZ. La temperatura y presión usada fue de 300 K y 1 atm, respectivamente. El método de Berendsen fue utilizado para controlar la temperatura. La constante de acoplamiento para el termostato de Berendsen fue de 0.1 ps [36].
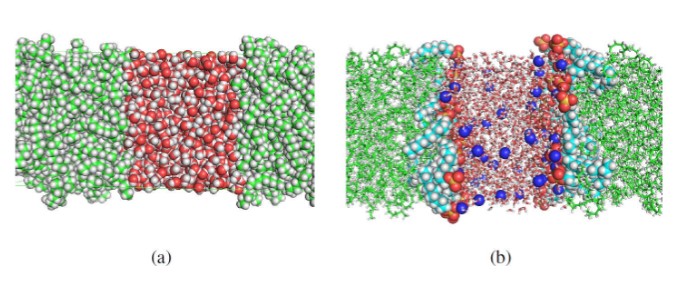
(a) Sistema agua/n-octano. (b) Sistema agua/SDS/n-octano. Ambos sistemas previamente relajados utilizando el programa Gromacs-4.5.5.
Las velocidades iniciales de las partículas fueron generadas usando una distribución Maxweliana a 300 K y las ecuaciones de movimiento se integraron usando el algoritmo leapfrog con un paso del tiempo de 1 fs. Las interacciones de Lennard-Jones se calcularon usando un radio de interacción de 1.40 nm y las interacciones electrostáticas fueron calculadas usando el procedimiento de mallado de Ewald (PME) con un radio de interacción de 1.30 nm [37].
Los sistemas vacío/SDS/agua/SDS/vacío fueron relajados usando el método de minimización gradiente conjugado. Luego, la configuración final obtenida se le aplicaron dos simulaciones de dinámica molecular tipo NVT. Las simulaciones tuvieron un lapso de 10 ns a 300 K. Con las trayectorias obtenidas de la segunda simulación tipo NVT, se estimó la tensión interfacial, la energía de formación de la interfaz (EFI) y el espesor de la película interfacial de cada uno de los sistemas. Para calcular el espesor de película interfacial se utilizó el criterio 10-90 por ser el más simple. Las trayectorias fueron almacenadas cada 1000 fs para determinar los promedios de las propiedades.
Los sistemas agua/n-octano y agua/sds/n-octano fueron relajados usando el método de minimización gradiente conjugado y steep descent. Las monocapas de surfac- tante fueron colocadas sobre las superficies del agua y estos sistemas SDS/agua/SDS fueron relajados usando una dinámica molecular tipo NVT de 500 ps. A las configuraciones finales obtenidas, se le colocaron las capas de n-octano en ambos lados de la celda. Estos sistemas fueron relajados haciendo una minimización con el método steep descent. Luego, la configuración final obtenida fue la usada para comenzar la dinámica molecular. Después del proceso de minimización, se realizaron simulaciones de dinámica molecular tipo NPT de 10 ns a 300 K. Posteriormente, se realizó una segunda simulación tipo NVT de 10 ns a las mismas condiciones de temperatura. Finalmente, se almacenaron las trayectorias cada 1 ps. Los últimos 5 ns de la dinámica molecular tipo NVT se utilizaron para determinar las propiedades.
La tensión interfacial fue estimada con el modelo de Kirwood-Buff usando los tensores de presión local y el espesor de película interfacial se determinó usando el criterio 10-90 y 90-90.
Resultados y Discusión
Perfiles de densidad de los sistemas vacío/SDS/agua
Inicialmente, con las trayectorias obtenidas de las dinámicas molecular tipo NVT, se determinaron los perfiles de densidad de los sistemas vacío/SDS/agua. En la figura 3, se muestran los perfiles de densidad de los sistemas agua/SDS/vacío simulados.
Como era de esperarse, se obtuvo un incremento en el espesor del perfil de densidad del SDS en el vacío a medida que aumenta el número de moléculas de SDS en la monocapa. Este comportamiento se debe a la mayor presencia de cadenas lipofílicas en los sistemas estudiados.
A su vez, se observa un solapamiento entre los perfiles de densidad del agua y el SDS, lo cual es debido a las interacciones tipo ión-dipolo de los iones sodio y el grupo hidrofílico sulfato presente en el surfactante con las moléculas de agua ubicadas en la región interfacial.
Adicionalmente, el perfil de densidad del agua se muestra invariable en el seno del líquido. También se observa que el perfil de densidad del agua se distorsiona a medida que aumenta la concentración de SDS. Este fenómeno ocurre en la región interfacial debido al aumento de las interacciones moleculares entre el SDS y el agua. En este trabajo, el sistema vacío/SDS/agua constituido por monocapas con 20 moléculas de SDS representa un superficie de agua saturada con dicho surfactante. Luego, aplicando el criterio 10-90 sobre los perfiles de densidad del agua en los sistemas vacío/SDS/agua, se determinaron los espesores de película interfacial. Estos valores se muestran en la tabla 1.
Para el sistema saturado vacío/SDS/agua, el espesor de película interfacial fue de 9.45 Á (tabla 1).
Esta magnitud de espesor de película interfacial se debe al aumento en las interacciones de la parte hidrofílica del SDS con el agua en la región interfacial. Si suponemos que el espesor de película del agua pura es de 4.5 Á, entonces el espacio ocupado por una monocapa de SDS en un sistema saturado es de 4.95 Á para el sistema vacío/SDS/agua. De igual manera, el espesor de película interfacial aumenta en función de la cantidad de moléculas de SDS en la región interfacial.
Tensión interfacial y energía de formación de la interfaz de los sistemas vacío/SDS/agua.
Para este trabajo se estimó la tensión interfacial de los sistemas vacío/SDS/agua en función del área ocupada por molécula de surfactante. Esto se puede apreciar en la figura 4.
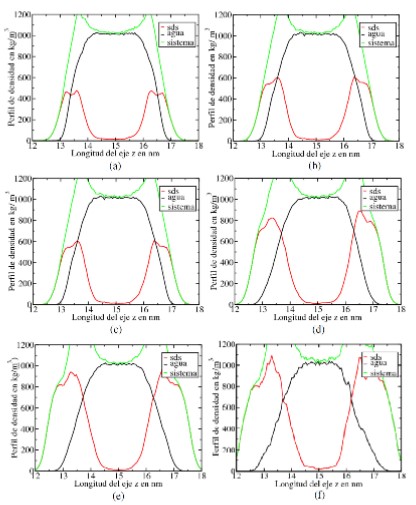
Perfiles de densidad de los sistemas SDS/agua/SDS. (a) 9 moléculas de SDS. (b) 12 moléculas de SDS. (c) 16 moléculas de SDS. (d) 20 moléculas de SDS. (e) 25 moléculas de SDS. (f) 30 moléculas de SDS.
| Moléculas de SDS en la monocapa Área por molécula en Á2 Película interfacial en Á | ||
| 9 | 100.00 | 7.42 |
| 12 | 75.00 | 7.88 |
| 16 | 56.25 | 8.45 |
| 20 | 45 | 9.45 |
| 25 | 36 | 10.46 |
| 30 | 30 | 11.12 |
Espesores de película interfacial de los sistemas vacío/SDS/agua/SDS/vacío obtenidos usando dinámica molecular tipo NVT a 300 K.
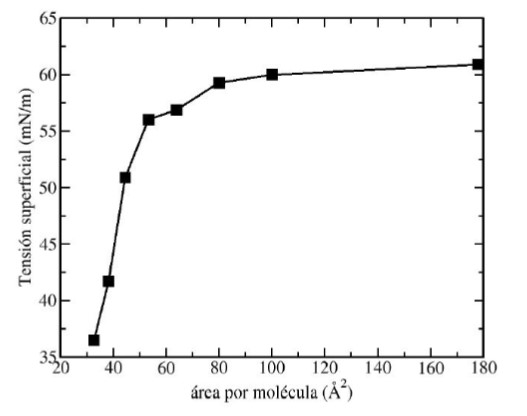
Valores de tensión interfacial en función del área por molécula de la monocapa de SDS.
Aquí, se encontró que cuando el área por molécula es mayor a 50 Á , la tensión interfacial de los sistemas vacío/SDS/agua se hace constante. En cambio, cuando el área interfacial es menor a 50 Á2por molécula se encuentra un cambio de pendiente en la curva de tensión superficial en función del área por molécula. Esos resultados muestran que la superficie del agua esta saturada con SDS cuando el valor de área superficial es de 50 Á2 . En cambio, cuando el área por molécula es muy grande, la tensión interfacial tiende al valor del agua pura. Para calcular el área por molécula se utilizá el método de regresión lineal sobre las curvas de tensión superficial y presión superficial de los sistemas vacío/SDS/agua. Para calcular la presión superficial, n, se utilizá la ecuación 7.
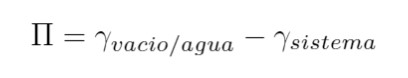 (7)
(7)Donde, es la tensión del agua pura yla tensión del sistema vacío/SDS/agua. En las figuras 5(a) y 5(b), se muestran las curvas de tensión interfacial y presión superficial en función del área por molécula para el sistema vacío/SDS/agua.
En este estudio usando dinámica molecular y el cambio de pendiente de la curva de presión superficial, se obtuvo un valor teórico de área por molécula de 53.3 Å2. En cambio, usando la curva de tensión superficial en función del área por molécula se obtuvo un valor de 54.3Å2. Usando medidas experimentales de tensión interfacial en agua fue reportado un valor de área por molécula para el SDS de 53 Å2[2]. Esto indica que la metodología aplicada es consistente para predecir las propiedades interfaciales del surfactante SDS en agua. A su vez, es importante destacar que la selección de un modelo de energía potencial con buenos parámetros moleculares permite predecir adecuadamente las propiedades interfaciales de los sistemas simulados.
Adicionalmente, se determiná la energía para la formación de la interfaz (EFI) de los sistemas vacío/SDS/agua. Para ello, se construyeron dos sistemas moleculares. El primer sistema fue construído con una molécula de SDS aislada en una celda periódica de dimensiones 4x4x30 nm3 . Luego, el segundo sistema consistía de una capa con 1000 moléculas de agua ubicada en el fondo de una celda periódica con las mismas dimensiones. A los dos sistemas se les realizaron dinámicas moleculares tipo NVT de 2ns a 300 K para calcular la energía total del surfactante SDS aislado, (), y la energía total de la capa de agua, (). A su vez, la energía total del sistema vacío/SDS/agua, (), se calculó para las configuraciones obtenidas en cada tiempo t de la simulación. La ecuación 8, muestra como se calcula la energía necesaria para la formación de la interfaz por molécula de surfactante (EFI):
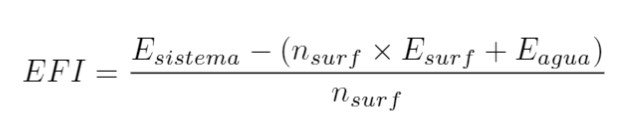 (8)
(8)Donde, nsurf, corresponde al número de moléculas de surfactantes presentes en la monocapa. La EFI se graficó en función del área por molécula de SDS. La figura 6, muestra la variación de dicha energía en función del área por molécula de SDS.
Cuando la superficie de agua se satura con moléculas de SDS, el sistema vacío/SDS/agua se hace más estable (figura 6). Por lo general, la energía de formación de la interfaz vacío/SDS/agua es una medida de las interacciones moleculares por surfactante que surge de la inserción de dichas moléculas en la región interfacial vacío/agua.
a) Tensión interfacial de los sistemas vacío/SDS/agua en función del área por molécula. b) Presión superficial de los sistemas vacío/SDS/agua en función del área por molécula.
Para un área por molécula de 80 Á2, el sistema muestra un cambio de pendiente. Aquí, la energía necesaria para la formación de la interfaz fue de -189.79 kJ/mol. Esto sugiere un aumento en las interacciones moleculares entre las moléculas de SDS que se acomodan sobre la superficie de agua. Dicha estabilización puede ser debida a las interacciones dispersivas entre las cadenas lipofílicas de los surfactantes que se acomodan sobre la superficie del líquido.
Interacción molecular entre el SDS y el agua.
En estos sistemas estudiados, es necesario entender el papel que juegan las interacciones moleculares entre el grupo hidrofílico y el agua en la reducción de la tensión superficial de los sistemas SDS/agua. Por tal motivo, usando la función de distribución radial entre átomos importantes del grupo hidrofílico y el agua, se estudio la interacción que existe entre la parte polar del surfactante SDS y las moléculas de agua. Para ello, se determinó la variación del número de moléculas de hidratación en función de la concentración de SDS ubicado en la región interfacial vacío/agua. En la figura 7, se muestra un ejemplo de la función de distribución radial obtenida para los sistemas vacío/SDS/agua.
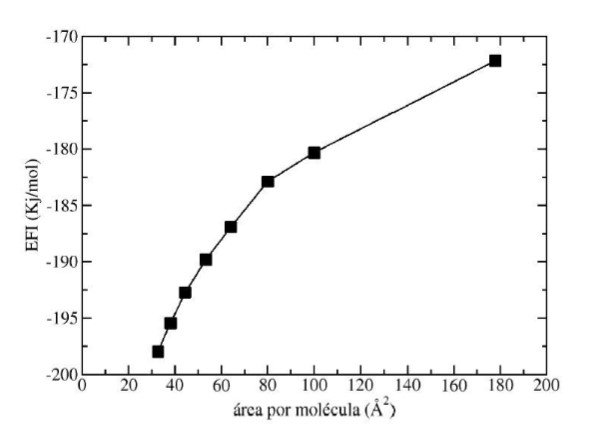
Energía para la formación de la interfaz de los sistemas vacío/SDS/agua en función del área por molécula.
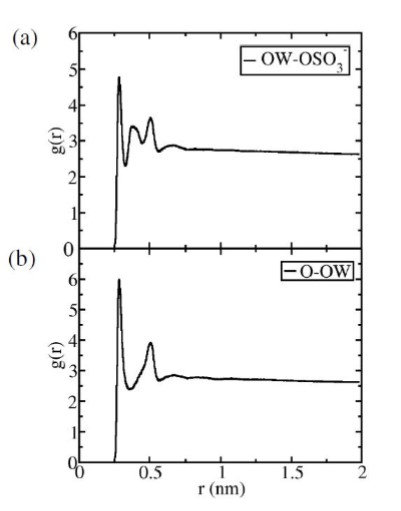
Función de distribución radial del sistema vacío/SDS/agua. Este sistema contiene 36 moléculas de SDS en la monocapa. a) OW representa el átomo de oxígeno del agua y OSO3 el grupo hidrofílico del surfactante. b) O representa los átomos de oxígenos del grupo hidrofílico y OW los del agua.
En la figura 7a, se muestra la función de distribución radial que corresponde a la interacción entre el grupo hidrofílico sulfato del surfactante SDS y las moléculas de agua. En este gráfico, se observa un primer pico que se presenta a una distancia r de 2.5 Á que corresponde a la primera capa de hidratación formada por las moléculas de agua. Seguidamente, se muestra otro pico con un máximo ubicado a una distancia r de 5 Á, el cual corresponde a la segunda capa de hidratación y sugiere que la diferencia de distancia entre los picos de la segunda y primera capa de hidratación corresponde a la de los puentes de hidrógeno que se forman entre las moléculas de agua. Entre los dos picos más grande, se presenta un pico de menor magnitud el cual descartamos en nuestro estudio porque estudiamos la formación de la primera capa de hidratatción.
El primer pico a una distancia r de 2.5 Á sugiere que se esta formando puentes de hidrógenos entre los oxígenos del grupo hidrofílico del surfactante y los átomos de hidrógeno que conforman a las moléculas de agua. Este fenómeno es lo que ocasiona la formación de la primera capa de hidratación alrededor del grupo hidrofílico del surfactante.
En la figura 7b, se muestra también la función de distribución radial obtenida usando la interacción de los oxígenos presentes en el grupo hidrofílico y el oxígeno de las moléculas de agua. El comportamiento obtenido es equivalente a la de la figura de arriba. La única diferencia que se presenta es que solo aparecen dos picos en este gráfico. De igual manera, el primero y segundo pico corresponden a la primera y segunda capa de hidratación, respectivamente. El primer pico aparece a 2.5 Á y esto sugiere que es indistinto hacer el análisis con cualquiera de los gráficos mostrados en la figura 7.
Generalmente, el número de moléculas de agua en la primera capa de hidratación es un parámetro efectivo para evaluar la interacción efectiva entre el grupo polar y el agua [3,38]. Esto indica que a mayor cantidad de moléculas de agua en la primera capa de hidratación, mayor es la interacción molecular entre el agua y el grupo hidrofílico[39].
Para calcular el número de moléculas de agua presentes en la primera capa de hidratación se utilizó la ecuación 9.
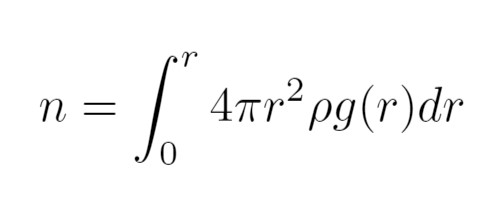 (9)
(9)Donde n denota el número de moléculas de agua en la primera capa de hidratación, r corresponde a la distancia hasta el primer pozo en la función de distribución radial y p es la densidad en número del agua en el bulk. En este trabajo, la densidad obtenidad fue de 100 moléculas de agua por volumen en nm3.
En la tabla 2, se muestran los valores de n obtenidos en función del número de moléculas de SDS presente en la monocapa de los sistemas vacío/SDS/agua.
Como el agua es una molécula netamente polar, la interacción entre el surfactante y el agua viene de la contribución de los átomos polares. En este caso, el número de moléculas aumenta en función del número de moléculas de surfactantes presentes en la monocapa. Como existen 4 átomos de oxígeno presente en el grupo hidrofílico del surfactante, se tiene que para la monocapa de 3x3 hay 4 moléculas de agua rodeando a cada átomo de oxígeno. En cambio, para el sistema 6x6 de surfactante se presentan 6 moléculas de agua por cada átomo de oxígeno presente en la monocapa.
En el punto de saturación de la superficie del agua con las moléculas de SDS, el número de moléculas de agua en la primera capa de hidratación corresponde a 19.74 moléculas. En ciertos trabajos [39,40], se han reportado un total de 18 moléculas de agua en la primera capa de hidratación para un sistema saturado con el SDS, lo cual indica que los resultados obtenidos en este trabajo son consistentes con los reportados en trabajos previos. A su vez, el modelo de energía potencial con el cual fue descrito el SDS es adecuado para desarrollar las simulaciones de dinámica molecular.
Perfiles de densidad de los sistemas agua/SDS/n-octano y agua/n-octano
En esta parte, se determinaron los perfiles de densidad de los sistemas agua/SDS/n-octano y agua/n-octano usando dinámica molecular tipo NPT a 300 K. Para la estimación de los espesores de película interfacial, se construyeron los perfiles de densidad usando 600 planos divisorios de la celda periódica a lo largo del eje z y la separación entre cada plano fue de 0,01 Á. En la figura 8, se muestra el perfil de densidad del sistema n- octano/agua obtenido después de la segunda simulación NPT de 5 ns a 300 K y 1 atm.
Los espesores de película interfacial se determinaron usando los criterios 10-90 y 90-90. El criterio 10-90 se refiere al aumento del perfil de densidad desde un 10 por ciento hasta un 90 por ciento y el criterio 90-90, corresponde al espesor medido entre los límites 90 por ciento y 90 por ciento de los perfiles de densidad de ambas capas inmiscibles. Usando el criterio 10-90, fueron obtenidos dos espesores de película interfacial usando las capas de hidrocarburo y agua.
Para el sistema agua/n-octano, los perfiles de densidad a lo largo del eje z son consistentes con las densidades experimentales de los líquidos puros. La densidad promedio obtenida para este sistema fue (820.84 ± 0.40) Kg/m3.
Generalmente, la densidad de los sistemas bifásicos oscila entre las densidades del agua y del hidrocarburo. En este trabajo, el modelo SPC usado para describir las moléculas de agua estima adecuadamente la densidaddel sistema agua/n-octano estudiado.
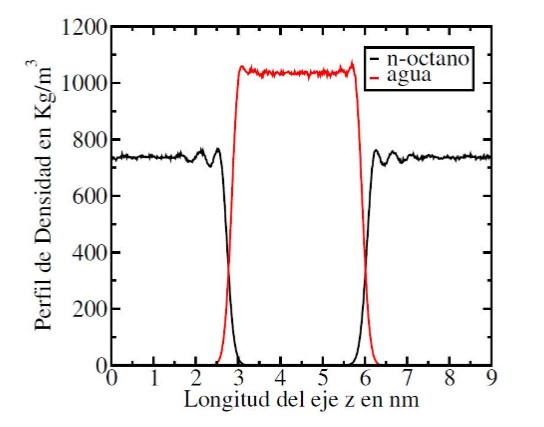
Perfil de densidad del sistema agua/n- octano obtenido con GROMACS 4.5.5.
Para el perfil del n-octano, se encuentran ciertas fluctuaciones de la densidad cerca de la región interfacial lo cual es debido al rearreglo de las moléculas de n-octano por la repulsión que presenta con las moléculas de agua. En cambio, el perfil de densidad del agua se encuentra sin perturbaciones debido a que el modelo usado en la simulación es muy rígido.
Usando el criterio 10-90, se encuentran dos valores de espesores de película interfacial usando los perfiles de densidad del agua y del n-octano. Con el perfil de densidad del agua, el espesor de película obtenido fue de 0.395 nm.
A su vez, utilizando el perfil de densidad del n-octano, el espesor de película interfacial fue de 0.415 nm. Estos resultados son consistentes con valores reportados ya mencionados anteriormente. Por ejemplo, Mitrinovic et al. reportaron un valor experimental de 0.350 nm para el sistema n-hexano/agua [41]. También, Riedleder et al. reportaron una amplitud interfacial de 0.380 nm para el sistema n-heptano/agua usando el criterio 10-90 [42]. En base a esto, encontramos que usando este criterio, los espesores de película interfacial son de mayor magnitud usando los perfiles de los hidrocarburos.
En cambio, usando el criterio 90-90, se encontró un único valor de espesor de película interfacial para los sistemas estudiados. En este caso para el sistema agua/n- octano, la magnitud fue de 0.495 nm. Generalmente, usando este criterio, los espesores de película interfacial son sobreestimados.
Luego, se colocaron monocapas de surfactantes SDS en la región interfacial del sistema agua/n-octano. En este caso, se estudio el efecto de la concentración de surfac- tante sobre esta propiedad interfacial denominada espesor de película. En la tabla 3, se muestran los valores obtenidos de espesor de película interfacial usando los criterios 10-90 y 90-90.
Se muestra que a medida que se adiciona moléculas de surfactantes a la región interfacial se genera un aumento de espesor de película interfacial. Usando el criterio 10-90 sobre la capa de agua, se encuentra magnitudes más coherentes de espesor de película interfacial. En cambio, usando la capa de n-octano hay un aumento considerable de los espesores de película. Lo mismo ocurre usando el criterio 90-90. Sin embargo, en todos los casos la película interfacial aumenta en función de la concentración de surfactante en la región interfacial (ver tabla 3). Desde el punto de vista teórico, el aumento de espesor de película implica una mayor estabilidad del sistema agua/SDS/n-octano. Generalmente, el SDS adicionado forma una monocapa autoensamblada en la región interfacial n-octano/agua y la parte lipofílica de este penetra la capa de hidrocarburo. A su vez, la parte hidrofílica interacciona fuertemente con el agua debido a la afinidad que presentan entre sí. Esto ocasiona la permeación del n-octano y agua en la membrana de surfactante autoensamblada y un aumento de los espesores de película medidos en las capas de n-octano y agua. De igual manera, con el criterio 90-90, se obtienen altos valores de espesor de película interfacial. De hecho, el aumento en el espesor de película en la capa de agua es debido al grupo hidrofílico. En cambio para la capa de hidrocarburo es debido a la cadena lipofílica.
En la tabla 4, se muestra el aumento en la película debido al grupo hidrofílico y lipofílico presentes en el surfactante dodecil sulfato de sodio. La contribución al espesor de película interfacial total debido al grupo hidrofílico es menor en comparación a la contribucióndel grupo lipofílico. A su vez, a medida que la concen-tración de surfactante aumenta la contribución de cadaparte del surfactante aumenta. Para el sistema comple-tamente saturado con surfactante, el cual corresponde a 36 moléculas en la monocapa, el espesor de película obtenido fue de 14.27 Å.
| MolÄl’culas de SDS | Criterio 10-90 en Á agua | Criterio 10-90 en Á n’octano | Criterio 90-90 en Á |
| 9 | 4.37 | 9.30 | 11.04 |
| 12 | 5.01 | 10.46 | 13.01 |
| 16 | 5.43 | 11.71 | 14.77 |
| 20 | 5.77 | 11.90 | 15.49 |
| 25 | 6.24 | 12.38 | 16.69 |
| 36 | 8.55 | 12.34 | 19.22 |
Espesores de película interfacial obtenidos para los sistemas agua/sds/n-octano usando los criterios 10-90 y 90-90.
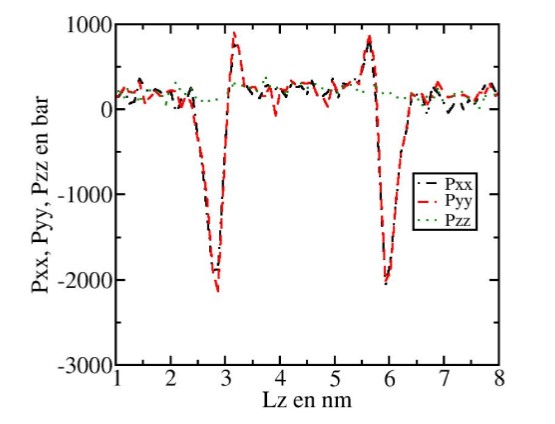
Tensores de presión local del sistema agua/n-octano obtenidos con GROMACS 4.5.5.
Tensión interfacial de los sistemas agua/SDS/n-octano y agua/n-octano
Las tensiones interfaciales de los sistemas agua/SDS/n- octano estudiados fueron obtenidas usando el modelo de Kirkwood-Buff. Para ello, se utilizan los tensores de presión local a lo largo del eje z de la celda periódica usada en la simulación (ver figura 9).
En la tabla 5, se muestran los valores obtenidos de nuestras simulaciones. Para el sistema n-octano/agua, la tensión interfacial obtenida por la simulación fue de 52.20 mN/m. El resultado obtenido en este trabajo es consistente con el valor experimental de 51.6 mN/m reportado por Zhang et al. [42].
Luego, al sistema agua/n-octano se le adicionó monocapas de surfactantes en la región interfacial para medir la capacidad reductora de la tensión interfacial que presenta el SDS y verificar que el modelo de energía potencial GROMOS53A6 describe adecuadamente a este tipo de moléculas.
La tensión interfacial disminuye a medida que aumentamos la concentración de surfactante SDS en la región interfacial. En nuestro caso, se obtuvo un mínimo de tensión interfacial cuando en la monocapas estaban presentes 36 moléculas de SDS. Usando esta cantidad de moléculas de surfactante, el área por molécula obtenido fue de 45.60 Á2el cual esta por debajo del valor experimental de 50.30 Á2. Esto garantiza que la región interfacial de la capa de agua este completamente saturada de surfactante y por lo tanto al usar los modelos de energía potencial GROMOS-53A6 y SPC se logró predecir la capacidad del SDS para reducir la tensión interfacial del sistema agua/n-octano usando dinámica molecular tipo NPT.
Conclusiones
En este trabajo, se lograron determinar las propiedades interfaciales de los sistemas vacío/SDS/agua y agua/SDS/n- octano usando dinámica molecular tipo NVT y NPT y los modelos de energía potencial GROMOS-53A6 y SPC para describir las moléculas de n-octano, SDS y agua, respectivamente.
El valor de área por molécula del SDS en agua estimado fue de 53.3 2 . A su vez, según la curva de energía necesaria para la formación de la interfaz se presenta un cambio de pendiente para un área de 80 2 y un valor de energía de -189.79 kJ/mol. Estos resultados muestran que las interacciones del SDS con el agua son efectivas cuando la superficie está saturada con dicho surfactante. A su vez, el espesor de la película interfacial aumenta en función de la concentración de surfactante SDS ubicado en la región interfacial.
Además, se encontró que el número de moléculas de agua presentes en la primera capa de hidratación aumenta en función del número de moléculas de SDS ubicadas en la región interfacial. En el punto de saturación de la superficie del agua, el número de moléculas de agua en la primera capa de hidratación corresponde a 19.74 moléculas.
También, se determinó la capacidad del SDS para reducir la tensión interfacial en un sistema agua/n-octano. Los perfiles de densidad del sistema agua/n-octano a lo largo del eje Z son consistentes con las densidades experimentales de los líquidos puros. La densidad promedioobtenida para este sistema fue (820.84±0.40)Kg/m3. Adicionalmente, para los sistemas agua/n-octano y agua/-sds/n-octano se pudo precisar que los espesores de película interfacial aumentan en función del número de moléculas de surfactantes presentes en la región interfacial con un mínimo de tensión interfacial de 4.94 mN/m y un valor máximo de espesor de película de 19.22 Å usando los force field GROMOS-53A6 y SPC.
| Moléculas de SDS | Grupo hidrofílico Á | Grupo lipofílico Á | Total Á |
| 9 | 0.42 | 5.15 | 6.09 |
| 12 | 1.06 | 6.31 | 8.06 |
| 16 | 1.48 | 7.56 | 9.82 |
| 20 | 1.82 | 7.75 | 10.54 |
| 25 | 2.29 | 8.23 | 11.74 |
| 36 | 4.6 | 8.19 | 14.27 |
Aumento de la película interfacial (a) en nm obtenidos usando los criterios 10-90 y 90-90. El agua fue descrita usando el modelo SPC-E.
| Moléculas de SDS | Tensión interfacial en mN/m | Error en mN/m |
| 9 | 50.30 | 0.42 |
| 12 | 47.41 | 1.31 |
| 16 | 42.15 | 0.21 |
| 20 | 35.41 | 0.50 |
| 25 | 27.68 | 0.39 |
| 36 | 4.94 | 0.30 |
Aumento de la película interfacial (a) en nm obtenidos usando los criterios 10-90 y 90-90. El agua fue descrita usando el modelo SPC-E.
Particularmente, estos resultados son debidos a la buena interacción que existe entre el grupo hidrofílico del surfactante y las moléculas de agua, lo cual genera una disminución de la tensión interfacial y un aumento del espesor de película.
Los resultados obtenidos de las simulaciones fueron consistentes con valores obtenidos experimentalmente.
Referencias
Myers, D. (2002). Surface activity and surfactant structures. Surfaces, Interfaces, and Colloids: Principles and Applications, Second Edition, 21-39.
Rosen, M. J., & Kunjappu, J. T. (2012). Surfactants and interfacial phenomena. John Wiley & Sons.
Myers, D. (2005). Surfactant science and technology. John Wiley & Sons.
Holmberg, K. (Ed.). (2002). Handbook of applied surface and colloid chemistry (Vol. 1). New York: Wiley
Rehfeld, S. J. (1967). Adsorption of sodium dodecyl sulfate at various hydrocarbon-water interfaces. Physical Chemistry, 71(3), 738-745, DOI: 10.1021/j100862a039.
Saien, J. & Akbari, S. (2006). Interfacial Tension of Toluene + Water + Sodium Dodecyl Sulfate from (20 to 50) ◦C and pH between 4 and 9. Chem. Eng. Data, 51(5), 1832-1835, DOI: 10.1021/je060204g.
Hansen, M. & Short, D. (1990). Optimization study of octane-in-water emulsions by sedimentation field-flow fractionation. Chromatography, 517, 333-344.
Tummala, N. R., & Striolo, A. (2008). Role of counterion condensation in the self-assembly of SDS surfactants at the water-graphite interface. Physical Chemistry B, 112(7), 1987-2000.
Watry, M. R., & Richmond, G. L. (2000). Comparison of the adsorption of linear alkanesulfonate and linear alkylbenzenesulfonate surfactants at liquid interfaces. American Chemical Society, 122(5), 875-883.
Zhao, T., Xu, G., Yuan, S., Chen, Y., & Yan, H. (2010). Molecular dynamics study of alkyl benzene sulfonate at air/water interface: effect of inorganic salts. Physical Chemistry B, 114(15), 5025-5033.
Shi, L., Tummala, N. R., & Striolo, A. (2010). C12E6 and SDS surfactants simulated at the vacuum-water interface. Langmuir, 26(8), 5462-5474.
Yan, H., Guo, X. L., Yuan, S. L., & Liu, C. B. (2011). Molecular dynamics study of the effect of calcium ions on the monolayer of SDC and SDSn surfactants at the vapor/liquid interface. Langmuir, 27(10), 5762-5771.
Shi, L., Tummala, N. R., & Striolo, A. (2010). C12E6 and SDS surfactants simulated at the vacuum-water interface. Langmuir, 26(8), 5462-5474.
Bresme, F., Chacón, E., Martínez, H., & Tarazona, P. (2011). Adhesive transitions in Newton black films: A computer simulation study. Chemical physics, 134(21), 214701.
Jang, S. S., & Goddard, W. A. (2006). Structures and properties of newton black films characterized using molecular dynamics simulations. Physical Chemistry B, 110(15), 7992-8001.
Bruce, C. D., Senapati, S., Berkowitz, M. L., Perera, L., & Forbes, M. D. (2002). Molecular dynamics simulations of sodium dodecyl sulfate micelle in water: the behavior of water. Physical Chemistry B, 106(42), 10902-10907.
Bruce, C. D., Berkowitz, M. L., Perera, L., & Forbes, M. D. (2002). Molecular dynamics simulation of sodium dodecyl sulfate micelle in water: micellar structural characteristics and counterion distribution. Physical Chemistry B, 106(15), 3788-3793.
Oostenbrink, C., Villa, A., Mark, A. E., & Van Guns- teren, W. F. (2004). A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force field parameter sets 53A5 and 53A6. Computational chemistry, 25(13), 1656-1676.
Malde, A. K., Zuo, L., Breeze, M., Stroet, M., Poger, D., Nair, P. C., ... & Mark, A. E. (2011). An automated force field topology builder (ATB) and repository: version 1.0. Chemical theory and computation, 7(12), 4026-4037.
Hermans, J., Berendsen, H. J., Van Gunsteren, W. F., & Postma, J. P. (1984). A consistent empirical potential for water-protein interactions. Biopolymers 23(8), 15131518.
Geysermans, P., Elyeznasni, N., & Russier, V. (2005). Layered interfaces between immiscible liquids studied by density-functional theory and molecular-dynamics simulations. Chemical physics, 123(20), 204711.
Rowlinson, J. S., & Widom, B. (2013). Molecular theory of capillarity. Courier Corporation.
Kirkwood, J. G., & Buff, F. P. (1949). The statistical mechanical theory of surface tension. Chemical Physics, 17(3), 338-343.
Irving, J. H., & Kirkwood, J. G. (1950). The Statistical Mechanical Theory of Transport Processes. IV. The Equations of Hydrodynamics. Chemical Physics, 18, 817-829.
Irving, J. H., & Kirkwood, J. G. (1950). The statistical mechanical theory of transport processes. IV. The equations of hydrodynamics. Chemical physics, 18(6), 817829.
Walton, J. P. R. B., & Gubbins, K. E. (1985). The pressure tensor in an inhomogeneous fluid of non-spherical molecules. Molecular Physics, 55(3), 679-688.
Neyt, J. C., Wender, A., Lachet, V., Ghoufi, A., & Mal- freyt, P. (2014). Quantitative Predictions of the Interfacial Tensions of LiquidaASLiquid Interfaces through Atomistic and Coarse Grained Models. Chemical theory and computation, 10(5), 1887-1899.
Van Gunsteren, W. F., & Berendsen, H. J. C. (1987). Groningen molecular simulation (GROMOS) library manual. Biomos, Groningen, 24(682704), 13.
Oostenbrink, C., Villa, A., Mark, A. E., & Van Guns- teren, W. F. (2004). A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force field parameter sets 53A5 and 53A6. Computational chemistry, 25(13), 1656-1676.
Bruce, C. D., Senapati, S., Berkowitz, M. L., Perera, L., & Forbes, M. D. (2002). Molecular dynamics simulations of sodium dodecyl sulfate micelle in water: the behavior of water. Physical Chemistry B, 106(42), 10902-10907.
Bruce, C. D., Berkowitz, M. L., Perera, L., & Forbes, M. D. (2002). Molecular dynamics simulation of sodium dodecyl sulfate micelle in water: micellar structural characteristics and counterion distribution. Physical Chemistry B, 106(15), 3788-3793.
Hess, B., Kutzner, C., Van Der Spoel, D., & Lindahl, E. (2008). GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. Chemical theory and computation, 4(3), 435-447.
Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., & Berendsen, H. J. (2005). GRO- MACS: fast, flexible, and free. Computational chemistry, 26(16), 1701-1718.
Lindahl, E., Hess, B., & Van Der Spoel, D. (2001). GROMACS 3.0: a package for molecular simulation and trajectory analysis. Molecular modeling annual, 7(8), 306-317.
Berendsen, H. J., van der Spoel, D., & van Drunen, R. (1995). GROMACS: a message-passing parallel molecular dynamics implementation. Computer Physics Communications, 91(1), 43-56.
Berendsen, H. J., Postma, J. V., van Gunsteren, W. F., DiNola, A. R. H. J., & Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. Chemical physics, 81(8), 3684-3690.
Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., & Pedersen, L. G. (1995). A smooth particle mesh Ewald method. The Chemical physics, 103(19), 8577-8593.
Zhao, T., Xu, G., Yuan, S., Chen, Y., & Yan, H. (2010). Molecular dynamics study of alkyl benzene sulfonate at air/water interface: effect of inorganic salts. Physical Chemistry B, 114(15), 5025-5033.
Xu, J., Zhang, Y., Chen, H., Wang, P., Xie, Z., Yao, Y., ... & Zhang, J. (2013). Effect of surfactant headgroups on the oil/water interface: An interfacial tension measurement and simulation study. Molecular Structure, 1052, 50-56.
Chen, Y., & Xu, G. (2013). Improvement of Ca 2+- tolerance by the introduction of EO groups for the anionic surfactants: Molecular dynamics simulation. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 424, 26-32.
Mitrinovic, D. M., Tikhonov, A. M., Li, M., Huang, Z., & Schlossman, M. L. (2000). Noncapillary-wave structure at the water-alkane interface. Physical review letters, 85(3), 582.
Riedleder, A. J., Kentish, S. E., Perera, J. M., & Stevens, G. W. (2007). Structural Investigation of a Water/n- Heptane Interface: A Molecular Dynamics Study. Solvent Extraction and Ion Exchange, 25(1), 41-52.
Zhang, Y., Feller, S. E., Brooks, B. R., Pastor, R. W. (1995). Computer simulation of liquid/liquid interfaces. I. Theory and application to octane/water. Chem. Phys., 23, 10252-10266.