

Estudio preliminar de la microbiota foliar de banano y su variación en presencia del patógeno Sigatoka Negra (Pseudocercospora fijiensis)
Preliminar study on the foliar microbiome in banana and its variation in presence of Black Sigatoka pathogen (Pseudocercospora fijiensis)
ACI Avances en Ciencias e Ingenierías
Universidad San Francisco de Quito, Ecuador
Recepción: 15 Mayo 2021
Aprobación: 09 Marzo 2022
Resumen: Se describe el perfil taxonómico bacteriano y fúngico de la hoja de banano (Musa x paradisiaca) en presencia y ausencia de la enfermedad Sigatoka Negra (Pseudocercospora fijiensis), evaluando manejos agronómicos orgánico y convencional en la provincia de El Oro, Ecuador. Las muestras recolectadas se sometieron a secuenciamiento de ADN y análisis en las regiones 16S (V3-V4) e ITS. Se observó que la microbiota fúngica de las hojas de banano del cultivo orgánico disminuyen su diversidad en presencia del patógeno, mientras que en el sistema convencional la diversidad aumenta. Además, se observa un amplicon sequence variant (ASV) del género Pseudomonas en mayor abundancia en la hoja sana orgánica. Este ASV proviene del clado de Pseudomonas fluorescens, un taxón conocido como benéfico para las plantas. La microbiota endófita presente en la filósfera del banano depende del sistema de cultivo y la presencia del patógeno cambia significativamente la composición microbiana.
Palabras clave: necrótico, secuenciamiento, diversidad, ASV, filósfera, diversidad, patógeno.
Abstract: We describe the bacterial and fungal microbiome of the banana (Musa x paradisiaca) leaf in healthy and necrotic phases of Black Sigatoka or black leaf streak disease (Pseudocercospora fijiensis), evaluated under organic and conventional agronomic management in El Oro province, Ecuador. Samples were collected for subsequent DNA sequencing and analysis of the 16S (V3-V4) and ITS markers. This study suggests that the fungal diversity from the organic farming system decreased due to the pathogen’s presence, while in the conventional farming system the diversity increased. Also, we observed that an amplicon sequence variant (ASV) from the genus Pseudomonas was more abundant in the healthy organic leaf. This ASV corresponded to the Pseudomonas fluorescens clade, a known beneficial taxon for plants. The banana’s phyllosphere endophyte microbiome depends on the cultivation system, and diseased conditions significantly change the microbiome in the presence of black leaf streak disease.
Keywords: necrotic, sequencing, diversity, ASV, phyllosphere, diversity, pathogen.
INTRODUCCIÓN
Las plantas son organismos multicelulares que proveen diversos ambientes o nichos ecológicos que son ocupados por una amplia gama de microorganismos [1], estos, a su vez forman asociaciones complejas y presentan diferentes roles en el desarrollo del hospedador entre ellos propiedades patogénicas, beneficiosas y algunos pueden no presentar un efecto directo sobre su hospedador [2]. Esto ocurre en los órganos situados bajo el suelo, al igual que los que se encuentran en la superficie. La filósfera hace referencia a las regiones aéreas de la planta, que en su mayoría son hojas [3]. En ellos habitan microorganismos en la superficie, conocidos como epífitos, así como dentro del tejido de la hoja, llamados endófitos [2].
La microbiota hace referencia al ensamblaje de microorganismos presente en un ambiente definido [4]. Ensayos de microorganismos cultivo-independientes, realizados en su mayoría en la última década, han permitido descifrar la composición taxonómica del microbioma asociado a plantas en diferentes estados y bajo varios estímulos [5]. El ensamblaje microbiano revelado en estos estudios no ocurre al azar [3], al contrario, se ha determinado que es producto de una selección derivada, principalmente, del genotipo de la planta y, posteriormente, de otros factores como estímulos bióticos y abióticos [6]. A niveles taxonómicos amplios la composición microbiana de la endósfera de la hoja presenta patrones similares entre especies y esto se diferencia significativamente del resto de la planta [7]. La comunidad bacteriana se encuentra dominada por los filos Proteobacteria y Firmicutes, mientras que la vasta mayoría de hongos que colonizan la endósfera provienen de los filos Ascomycota y Basidiomycota [1].
Datos explorados anteriormente sugieren que los microorganismos con la capacidad de colonizar la endósfera de la hoja tienen la necesidad de poseer motilidad, la habilidad de degradar la pared celular vegetal y activar en el huésped respuestas inmunitarias [7]. Se conoce que en las raíces cuando hay deficiencia de nutrientes específicos el estado inmunológico de la planta se reprime y esta recluta microorganismos creando un ensamblaje microbiano específico que incrementa la biodisponibilidad de los nutrientes en deficiencia [8, 9], moviliza los nutrientes dentro del hospedador y además puede cambiar el metabolismo de la planta mediante la excreción de moléculas para redireccionar la energía generada a otros destinos metabólicos [10].
Así mismo, la presencia de comunidades heterogéneas en la endósfera del hospedador le generan en este último modificaciones metabólicas que le confieren resistencia a patógenos además de secretar sustancias antimicrobianas que los repelen [11]. Ensamblajes microbianos específicos confieren tolerancia al estrés abiótico [12]. En conjunto, el microbioma puede modificar el proceso evolutivo de una especie incrementando el fitness de la planta. En este contexto, se considera al microbioma asociado a un organismo, como un genoma extendido que juega un rol fundamental en el desarrollo del hospedador [13, 14]. Estas propiedades están bien caracterizadas en las comunidades microbianas asociadas a la raíz, tanto en la endósfera como en la rizósfera, sin embargo, en la región superior de la planta los estudios son escasos y actualmente existen muy pocos ensayos que demuestran el efecto de la microbiota de la filósfera [15, 16].
La disbiosis es la condición en la que existe un desbalance en la microbiota y este se encuentra asociado a un fenotipo enfermo [16], en varios estudios se ha encontrado que en presencia de un patógeno existe un drástico aumento de la población del filo Proteobacteria y disminución de Firmicutes [6, 16-18], además de la disminución en la diversidad [16, 17, 19]. Estos hallazgos abren puertas al desarrollo de tecnologías basadas en ensamblajes microbianos sanos que permitan revertir el estado disbiótico y le devuelvan el estado sano al hospedador [18].
El banano (Musa spp.) se encuentra entre los productos de mayor interés agronómico desde el punto de vista económico. Ecuador es el mayor exportador abarcando el 34% del mercado [20]. A nivel mundial los cultivos de banano Cavendish se ven afectados por el patógeno foliar más devastador para estas plantaciones Pseudocercospora fijiensis, antes conocido como Mycosphaerella fijiensis, o comúnmente denominado Sigatoka Negra, es un hongo Ascomyceto hemibiotrófico heterotálico [21]. Actualmente, el único manejo fitosanitario en plantaciones afectadas es la aplicación semanal de fungicidas sintéticos [22]. Lo que representa un porcentaje considerable de los costos de producción, además de tener como consecuencia impactos negativos a nivel ecológico y de salud pública [21, 23].
El objetivo del presente estudio es la caracterización taxonómica de la microbiota endófita de la hoja de banano en presencia y ausencia de sintomatología de Sigatoka Negra (P. fijiensis) contrastando manejo orgánico y convencional en la provincia de El Oro, Ecuador. Este estudio explora las poblaciones microbianas de la hoja de banano encontrando posibles controladores biológicos para el patógeno de interés.
MATERIALES Y MÉTODOS
Recolección de muestras
En el mes de mayo del 2019 se recolectaron muestras de hojas de banano de dos plantaciones localizadas en el cantón Santa Rosa, provincia de El Oro, (Fig. 1). La primera estuvo ubicada en la hacienda “Central, San José”, con coordenadas 3° 21’ 18.000” N y 79° 54’ 5.800” E, con un manejo fitosanitario orgánico usando controladores biológicos, compostaje y fertilizantes orgánicos. La segunda plantación estuvo ubicada en la hacienda“Cueva”, cuyas coordenadas son 3° 21’ 33.700”N y 79° 54’ 58.300” E, y se encuentra bajo manejo convencional usando fungicidas e insecticidas sintéticos de manera rotatoria, además de fertilizantes sintéticos. Se seleccionó el tercio medio de la cuarta hoja de cinco plantas que fueron muestreadas aleatoriamente para cada tratamiento. Con estas cinco muestras se realizó un pool para cada tratamiento, los tratamientos son: plantas con sintomatología de Sigatoka Negra y plantas asintomáticas, esto para cada plantación (orgánico y convencional). En el campo se realizó un lavado con una solución estéril compuesta por bifosfato de sodio 45,9 mM, fosfato disódico 61,6 mM y 0,1 % Tween 20. Posteriormente, se conservó a 4°C para su transporte al laboratorio [24].

Plantaciones de banano (Musa x paradisiaca) en la localidad Santa Rosa, El Oro. (A) Hacienda “San José” con sistema de cultivo orgánico, (C) hoja sana, (D) hoja enferma, (G) acercamiento a la hoja sana y (H) a la hoja enferma. (B) Hacienda “El Cueva” con sistema de cultivo convencional, (E) hoja sana, (F) hoja enferma, (I) acercamiento a la hoja sana y (B) a la hoja enferma.
Desinfección y conservación
Una vez en el laboratorio, las hojas se cortaron en secciones pequeñas con el fin de facilitar su procesamiento. Se desinfectó la superficie foliar, dentro de una cámara de flujo laminar manteniendo condiciones de asepsia, con una solución de hipoclorito de sodio al 10 % y Tween al 0,1 % durante dos minutos. Luego se eliminó los residuos del desinfectante en agua destilada estéril durante dos minutos adicionales. Posteriormente, se dejaron secar las muestras en papel estéril [24]. Cuando las muestras se secaron se las congeló con nitrógeno líquido previo a liofilizarlas, con el fin de conservar la población de endófitos.
Recuperación de endófitos
Para la realización de la digestión enzimática se tomó 0,5 g de tejido seco pulverizado de cada planta (pool de 5g por tratamiento). A cada pool se añadió 25 mL (relación 5:1 buffer) de buffer de digestión compuesto por Macerozyme al 0,1 %, celulasa al 1 %, D-Manitol 0,7 M, MES 5 mM, cloruro de calcio di-hidratado 9 mM, fosfato diácido monobásico 65 uM ajustado a un pH de 5,7. Se incubó en un shaker a 28°C, 125 rpm, durante 12 horas [25].
Pasadas las 12 horas de digestión del tejido foliar se realizó dos centrifugaciones seriadas a 200 rpm durante cinco minutos, rescatando el sobrenadante y evitando incluir restos de tejido foliar. La última centrifugación se llevó a cabo a 3000 rpm durante veinte minutos, con el fin de rescatar los microorganismos endófitos presentes en la solución. El pellet se resuspendió en solución salina y se conservó a -20°C en tubos Eppendorf [25].
Extracción de ADN y secuenciamiento de microbioma
La extracción de ADN de hongos y bacterias endófitas recuperadas se realizó con el kit comercial DNeasy Powersoil® (Qiagen, Hilden, Alemania) usando el protocolo estándar establecido en el kit [23]. La concentración y el grado de pureza de las muestras se midieron usando Qubit 4 Fluorometer (Invitrogen, Thermo Fisher Scientific, USA).
Para bacterias se amplificó la región V3–V4 del gen 16S rRNA usando los primers 338F (5′-ACTCCTACGGGAGGCAGCA-3′) y 806R (5′-GGACTACHVGGGTWTCTAAT-3′). Se añadieron dos barcodes y 6 cambios en marcos de lectura en el extremo 5’ del primer 338F, y en el primer 806R basado en el protocolo. Cada reacción de PCR se realizó por triplicado y cada una incluye una mezcla única de 3 primers con cambio de marco para cada plato. El protocolo de PCR se realizó con 5 μl de Kapa Enhancer, 5 μl de Kapa Buffer A, 1,25 μl of, 5 μM de cada primer y 0,375 μl de una mezcla de PNAs, 0,5 μl de Kapa dNTPs, 0,2 μl Kapa Robust Taq (Kapa Biosystems, Wilmington, MA), 8 μl dH2O, 5 μl DNA. Las condiciones de temperatura fueron: 95°C durante 60 segundos, 24 ciclos de 95°C por 15 segundos, 78°C durante 10 segundos, 50°C por 30 segundos, 72°C por 30 segundos y finalmente, 4°C hasta su uso. Los amplicones se purificaron con beads magnéticos AMPure XP (Beckman Coulter, Brea, CA) y se cuantificaron mediante el uso del fluorómetro Qubit 2.0 (Invitrogen, Carlsbad, CA). El secuenciamiento se llevó a cabo usando 10 pM en el equipo Ilumina MiSeq (Ilumina, San Diego, CA) como describen Finkel y colaboradores [26].
En cuanto a la región ITS los primers usados son ITS1-F (5′- CTTGGTCATTTAGAGGAAGTAA-3′) e ITS2 (5′-GCTGCGTTCTTCATCGATGC-3′) (). Las muestras de ADN se diluyeron a concentraciones de 3,5 ng μl-1. Luego se prepararon reacciones por triplicado compuestas por 10 ng de ADN, 1 U de buffer, 0,3 % de suero bovino de albúmina, 2 mM de cloruro de magnesio, 200 μM de dNTPs, 300 nM de cada primer y 2 U de DFS-Taq DNA polimerasa (Bioron, Ludwigshafen, Alemania). Las condiciones del ciclo son 2 minutos a 94°C, 25 ciclos de 30 segundos a 94°C, 30 segundos a 55°C y 30 segundos a 72°C, finalmente, la terminación del protocolo se realizó a 72°C durante 10 minutos. El producto del PCR atravesó un proceso de purificación con exonucleasas. Además, se realizó un segundo protocolo de PCR con el fin de añadir los barcodes específicos para las muestras. Luego, se purificó los amplicones con el kit AMPure XP. Finalmente, se unieron las muestras en proporciones iguales y se diluyó a 10 pM para su posterior secuenciamiento. El secuenciamiento se llevó a cabo en el equipo Ilumina MiSeq usando el kit 600-cycle V3 [26].
Procesamiento de datos
Usando MT-Toolbox, se verifico que las secuencias obtenidas tengan el 100 % de la secuencia del primer de amplificación y se empalmaron entre sus respectivos pares [27]. Las secuencias resultantes se filtraron por calidad utilizando Sickle al no permitir ninguna ventana con una puntuación Q inferior a 20 [28]. Luego de este filtro, todas las muestras que poseían menos de 1000 reads fueron descartadas. Posteriormente, las secuencias resultantes se colapsaron en amplicon sequence variants (ASVs) utilizando el paquete R DADA2 versión 1.8.1. Por último, la asignación taxonómica de ASVs se llevó a cabo mediante el método naive Bayes kmer en DADA2, usando la base de datos Silva 132 [29]. Los ASVs hacen referencia a la recuperación de secuencias exactas presentes en la muestra, basándose en un modelo estadístico de error, por lo tanto, cada ASV corresponde a una única secuencia encontrada en la muestra correspondiente a un único taxón [30].
Por otro lado, las secuencias del ITS fueron procesadas con el paquete DADA, usando los parámetros default en los reads forward. La asignación taxonómica de los ASVs se realizó implementando el método naive Bayes kmer, usado en el paquete MOTHUR usando la base de datos UNITE [27, 31].
Análisis de datos
Previo al análisis de datos se normalizaron las muestras por medio de rarificación a 1000 reads. Las tablas de conteo resultantes se analizaron con los paquetes de R Ohchibi, ComplexHeatmap y Pheatmap. Los parámetros de α-diversidad y los filogramas se realizaron con el paquete Ohchibi [26]. Por otro lado, el paquete ComplexHeatmap se usó para realizar los UpSet plots que permitieron determinar el core microbiome, que en este caso se define como todos los microorganismos endófitos presentes en la hoja independientemente del estado de salud o sistema de manejo agronómico bajo el que se encuentre la planta; así como otros consorcios de ASVs en común entre muestras [32]. Además, el paquete Pheatmap se usó para realizar heatmaps que permitieron determinar de manera visual que ASVs tenían un mayor número de conteos en comparación a la media en cada muestra, basándose en el Z-score [33]. Finalmente, se realizó un árbol filogenético mediante el algoritmo Maximum Likelihood y modelo Tamura Nei para comparar los ASVs de interés con secuencias de la base de datos RDP para procariotas [4, 34].
α-diversidad y composición filogenética microbiana
En este estudio, se colectó y secuenció los datos de 4 pooles de las hojas de banano (Fig. 1). Se obtuvo un total de 52 717 conteos de la región 16S (V3-V4) y 575 599 conteos de la región ITS. Una vez realizado el análisis de las secuencias estos se clasifican en 410 ASVs bacterianos y 234 ASVs fúngicos. Las secuencias resultantes se encuentran depositados en la siguiente base de datos (https://github.com/darioxr/ACI_banano_sigatoka).
En la Figura 2 se presenta la abundancia relativa de la composición filogenética, en la sección (A) el filo bacteriano más abundante en ambas plantaciones es Proteobacteria con 77,67 % y 96,29 % de abundancia relativa en convencional y orgánico respectivamente. Se muestra baja abundancia de Firmicutes en general, siendo mayor en la hoja convencional con 21,08 %. La comunidad microbiana fúngica de la plantación orgánica muestra distribución uniforme, los órdenes Erotiales y Malasseziales son los más abundantes con 23,32 % y 25,23 % respectivamente. Por otro lado, la hoja de la plantación convencional se ve dominada, en su mayoría por los órdenes Cystobasidiales y Sporidiobolales con 53,89 % y 31,14 % respectivamente.
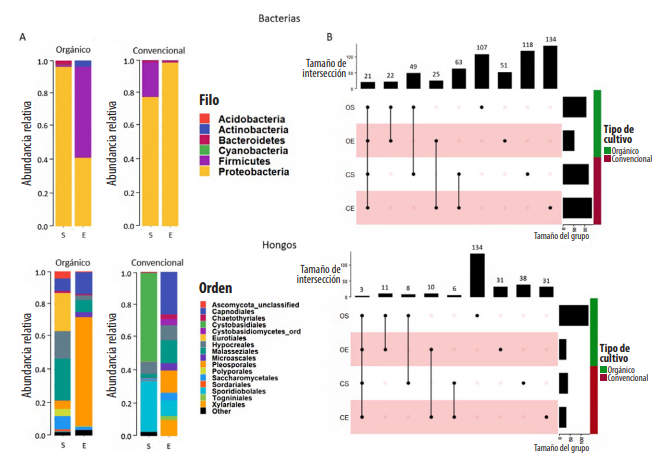
Cambio en (A) composición filogenética y (B) riqueza de ASVs en presencia de P. fijiensis para el gen 16S rRNA (V3-V4) y la región ITS: orgánico (O), convencional (C), sano (S) y enfermo (E).
La respuesta de la composición filogenética del microbioma bacteriano es opuesta para cada sistema de manejo, en términos de proporción de los filos Firmicutes y Proteobacteria. La comunidad fúngica del sistema de manejo orgánico se ve dominada por el orden Cystobasidiales en presencia del patógeno, mientras que el convencional se vuelve más uniforme. El microbioma core está compuesto por 21 ASVs bacterianos y 3 ASVs fúngicos. En cuanto a hongos, la hoja orgánica sana posee una riqueza mayor en comparación con las demás muestras.
Todos los parámetros de α-diversidad del microbioma bacteriano en la plantación orgánica disminuyen en presencia del patógeno (Tabla 1). Por otro lado, en la hoja sometida a manejo convencional los parámetros de α-diversidad en bacterias disminuyen en presencia del patógeno, con excepción del índice de Shannon y la uniformidad (evenness) que se mantienen en un rango similar. La comunidad fúngica, al igual que la bacteriana, disminuye su diversidad en la plantación orgánica, mientras que en la plantación convencional la α-diversidad aumenta en presencia del patógeno, con excepción de Chao1 que se mantiene similar.
| Organismo | Manejo agronómico | Estado | Shannon | Chao1 | InvSimpson | Evennes |
| Bacterias | Orgánico | Sano | 2,20 | 160,00 | 5,62 | 0,47 |
| Enfermo | 1,50 | 60,56 | 2,92 | 0,38 | ||
| Convencional | Sano | 1,95 | 235,44 | 4,22 | 0,41 | |
| Enfermo | 1,97 | 179,38 | 3,63 | 0,40 | ||
| Hongos | Orgánico | Sano | 3,52 | 134,00 | 16,50 | 0,71 |
| Enfermo | 1,63 | 31,00 | 2,22 | 0,47 | ||
| Convencional | Sano | 1,54 | 38,00 | 2,85 | 0,42 | |
| Enfermo | 3,21 | 31,00 | 22,00 | 0,93 |
α-diversidad según manejo agronómico en presencia de P. fijiensis
El microbioma bacteriano responde disminuyendo su diversidad en presencia del patógeno, mientras que el fúngico responde diferente dependiendo del sistema de manejo agronómico. En el microbioma de la hoja de banano orgánica disminuye la diversidad, mientras que en el convencional aumenta.
En cuanto a la composición filogenética que se muestra en la Figura 2, la comunidad bacteriana en la sección (A), la hoja orgánica muestra mayor abundancia de la población del filo Firmicutes de 0,83 % a 54,79 % y de Proteobacteria de 96,29 % a 41,38 % en presencia de P. fijiensis. Mientras que la hoja convencional muestra el efecto contrario, Proteobacteria domina la población cubriendo un 77,67 % en estado sano y llegando hasta 98,54 % al final de la infección. Por otro lado, en la población fúngica de la plantación orgánica el orden Pleosporales posee la mayor abundancia (66,30 %) en hojas que presentaron infección. Mientras que, en convencional, en estado de infección la población fúngica aunque presenta mayor uniformidad en su comunidad, el orden más abundante es Capnodiales.
CORE MICROBIOME
Existe una mayor riqueza de ASVs bacterianos en las muestras del manejo agronómico convencional incrementando el número de ASVs de sano a enfermo (Figura 3B). Mientras que el manejo orgánico muestra menor número de ASVs en general y estos disminuyen a alrededor de la mitad en presencia del patógeno. En este estudio se definió al core microbiome al conjunto de ASVs que se comparten independientemente del manejo agronómico y el estado de salud vegetal. La composición del core bacteriano se encuentra dominada por ASVs asignados al género Pantoea sp., varios ASVs de la familia Enterobacteriaceae y un ASV asignado a Pseudomonas sp. (Figura 3B). Se observa que la composición microbiana se diferencia principalmente dependiendo del estado de salud del huésped, es decir, sano o enfermo. Además, es posible identificar clusters exclusivos para cada tratamiento (Figura 3A). El “Cluster a” que se compone por dos ASVs del género Pantoea sp. se encuentran incrementados de manera exclusiva en la muestra orgánica enferma, el “Cluster b” compuesto por Enterobacterias y Pantoea sp. se incrementa en la muestra convencional enferma, además del “Cluster c.2” compuesto por diferentes ASVs con las mismas asignaciones taxonómicas. Por otro lado, el “Cluster c” es compartido por las muestras del manejo convencional siendo el “Cluster c.1” exclusivo para el estado sano. Además, las muestras orgánicas sanas poseen ASVs incrementados de los géneros Pantoea sp. y Pseudomonas sp. de manera exclusiva.
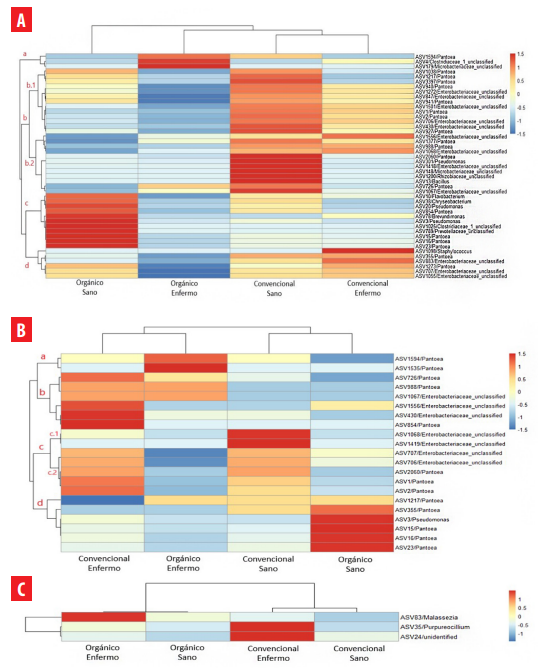
Heatmap ploteado con ASVs de (A) muestras bacterianas y el microbioma core (B) bacteriano de 21 ASVs y (C) fúngico de 3 ASVs. El gráfico muestra el enriquecimiento diferencial de ASVs dependiendo del estado de salud y el manejo agronómico.
Los ASVs bacterianos incrementan de manera diferencial dependiendo del sistema de manejo y del estado de salud de la hoja. Los ASVs incrementados en la muestra orgánica sana son de interés para prospección. El core bacteriano se muestra dominado por el género Pantoea sp. y géneros de la familia Enterobacteriaceae, mostrando clusters definidos para cada muestra y agrupándose por estado de salud. El core fúngico muestra agrupación por manejo agronómico.
La riqueza fúngica presente en las muestras pertenecientes a la hoja orgánica sana es más de cuatro veces mayor que la presente en las otras muestras (Figura 2B). Además, el core fúngico solo comprende a tres ASVs que pertenecen a los géneros Malassezia sp., Purpureocilium sp. y ASV no identificado (Figura 3C). Adicionalmente, los ASVs de las muestras sanas no se encuentran incrementados.
IDENTIFICACIÓN DE MICROORGANISMOS DE INTERÉS
La Figura 3 en la sección (A) muestra los ASVs bacterianos presentes en todas las muestras. Se observa que las muestras de la plantación orgánica presentan clusters definidos de escasos ASVs que se encuentran altamente enriquecidos sobre la media. Además, todos los ASVs que se encuentran sobre la media en el estado sano, disminuyen en presencia del patógeno y en este último estado tres ASVs correspondientes al género Pantoea sp. y dos géneros no clasificados pertenecientes a las familias Microbacteriaceae y Clostridiaceae incrementan sobre la media. Por otro lado, las hojas del cultivo convencional muestran un espectro más amplio de ASVs aumentados, tanto en sano como en el estado enfermo.
El cluster que posee ASVs enriquecidos en la hoja de la planta sana que se encontraba en la plantación orgánica es exclusivo de esta muestra, es decir, en ninguna de las otras muestras se observa enriquecimiento evidente de estos ASVs. En este contexto, la Figura 4 muestra el árbol filogenético creado para el ASV3 correspondiente al género Pseudomonas sp. enriquecido en la muestra sana orgánica, en él se observa que el ASV3 se ubica en el clado exclusivo de Pseudomonas fluorescens y se separa del clado de Pseudomonas syringae.
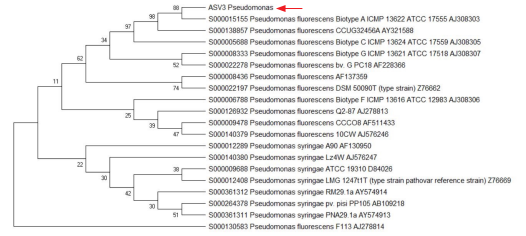
Árbol filogenético realizado con Maximum Likelihood entre el ASV3_Pseudomonas, P. fluorescens y P. syringae.
El árbol filogenético se realiza con el modelo Namura Tei y el método Maximum Likelihood usando un Bootstrap de 10000. El ASV3 de interés se ubica dentro del caldo de Pseudomonas fluorescens.
DISCUSIÓN
Los estudios de microbioma endófito de la filósfera son cada vez más informativos debido a recientes estudios que demuestran causalidad de un ensamblaje microbiano específico sobre efectos disbióticos en el fenotipo foliar de la planta [16]. Sin embargo, estos estudios en planta de banano son escasos y frente a diversos patógenos amenazando la seguridad alimentaria de la fruta más consumida a nivel mundial, existe un creciente interés en generar opciones que contribuyan a la sostenibilidad agronómica.
Los microbiomas en su estado basal entre sistemas de manejo agronómico presentan diferencias estructurales y de diversidad claras. Los resultados de α-diversidad de este estudio no son consistentes con la mayoría investigaciones anteriores. Sin embargo, existe otra evaluación en plantas de uva que demuestra mayor diversidad en una plantación orgánica, que en una convencional [36]. Sin embargo, se realizó un estudio similar en trigo que no demuestra diferencias significativas [36]. En composición filogenética, Proteobacteria corresponde al filo de bacterias más abundante en microbioma vegetal, independientemente de la región, el manejo de cultivo y de la fracción de la planta [1]. El aumento del filo Firmicutes en la plantación convencional se ha observado antes en la filósfera de maíz [37]. Por otro lado, a nivel de género, Pantoea corresponde al más común en la filósfera de las plantas [38].
Es necesario tomar en cuenta que los manejos agronómicos orgánicos pueden variar mucho en las prácticas aplicadas en el cultivo de una misma especie y en diferentes especies, por lo tanto, este factor es fundamental en la evaluación del microbioma. Este estudio considera como cultivo orgánico al conjunto de prácticas para el manejo integral de plagas que excluyen el uso de fungicidas de origen sintético. Se pretende comparar entre la microbiota de prácticas orgánicas, descritas anteriormente, y prácticas convencionales que usan fungicidas sintéticos. Una de las limitaciones del estudio se debe a que no se considera la influencia de las diversas prácticas, dentro de los cultivos orgánicos, en los cambios de la microbiota.
Se conoce que al entrar un patógeno las comunidades microbianas que residen en la endósfera de la hoja atraviesan cambios en cuanto a estructura y diversidad. En el caso de las bacterias, disminuye la diversidad y el patrón general de disbiosis es que la abundancia de Firmicutes disminuya y aumente en el filo Proteobacterias lo que ocurre en la plantación convencional, más no en la plantación orgánica [6, 16-18]. La respuesta observada en la plantación orgánica no se ha reportado anteriormente en estudios publicados de evaluaciones de microbioma foliar. Sin embargo, la disminución en la diversidad bacteriana en disbiosis ha sido reportada en varios ejemplos [16, 17, 19].
Aunque los hongos del microbioma vegetal, en general, han sido escasamente explorados, los pocos reportes existentes demuestran que Ascomicetos es el filo más abundante en la filósfera de plantas sanas y enfermas [19, 39-42], y la clase Dothideomycetes incrementa en presencia de un patógeno [19, 38]. Estudios previos que han estudiado el cambio en la diversidad de la hoja en presencia de un patógeno hemibiótrofo han encontrado una mayor diversidad al final de la infección, lo que también ocurre en la plantación convencional infectada con P. fijiensis en nuestro estudio [39, 40]. Sin embargo, en la plantación orgánica una vez necrosado el tejido la diversidad disminuye, lo que no se ha reportado anteriormente en la literatura.
Esto sugiere que realmente existen patrones de respuesta en la diversidad y composición filogenética que se ajustan más a lo que ocurre en la hoja de banano de una plantación convencional. Lo observado en las comunidades fúngicas de plantación orgánica no se ha reportado anteriormente, por lo tanto, es un punto de investigación interesante para prospección sobre microorganismos que probablemente están interviniendo de manera activa en el control sobre el desarrollo del patógeno.
Entre los microorganismos que se encuentran aumentados en la muestra de la planta orgánica sana se encuentra el género Pseudomonas sp. Este género comprende cientos de especies que poseen características como potenciadores de crecimiento y controladores biológicos mediante diversos mecanismos [43]. Sin embargo, dentro de este género también se encuentra una especie fitopatógena que en las últimas décadas se ha convertido en un modelo de estudio que ha llevado a la ciencia vegetal a comprender diversos patrones de patogénesis [44]. Existen varias cepas de la especie Pseudomonas fluorescens que han sido evaluadas como prometedoras en plantas de banano para el control de Fusarium oxysporum [45]. Desde entonces, se han realizado varias evaluaciones de esta especie como antagonistas a diversos fitopatógenos como F. oxysporum f. sp. ciceris (agente causal de la enfermedad Fusarium wilt), Rhizoctonia solani (agente causal de blight del arroz), Pyricularia oryzae (agente causal de blast del arroz), entre otras [46].
Pseudomonas fluorescens Pf1 es una cepa bacteriana que produce sideoforos, cianido de hidrógeno, antibióticos y pioluteorina, además induce la resistencia asociada a enzimas en raíces de plantas de banano para combatir Foc [47]. En base a estos reportes se han realizado diversos ensayos demostrando la efectividad de varios formulados y protocolos de aplicación de P. fluorescens Pf1 contra el patógeno de suelo F. oxysporum. Además, se conoce que la aplicación de formulados de esta cepa provee mayor protección a estrés ambiental a plantas de banano [46]. P. fluorescens Pf1 es un ejemplo de microorganismos que atraviesan procesos evolutivos de especialización con la planta huésped de banano para conferirle características que le provean beneficios para combatir la colonización del tejido vegetal por fitopatógenos. Hemos mencionado que el potencial de interacción planta microbioma en la región inferior, es decir, bajo la superficie de la tierra, ha sido ampliamente explorado [1]. Por esto, explorar y explotar el potencial del microbioma foliar que se encuentran en la región de infección en la planta interactuando directamente con el patógeno es un área muy interesante por explorar.
En este estudio, Proteobacterias se muestra como el filo más abundante en ambas plantaciones, con un ligero incremento del filo Firmicutes en la hoja perteneciente a convencional. En cuanto a hongos el filo más abundante es Ascomycota. De manera detallada, el microbioma bacteriano disminuye en términos de diversidad cuando P. fijiensis necrosa la hoja independientemente del sistema de cultivo. Por otro lado, la respuesta del microbioma fúngico frente al patógeno varía dependiendo del sistema de manejo agronómico. En el caso de la plantación convencional la diversidad aumenta al final de la infección, mientras que en el manejo orgánico la diversidad disminuye. El core microbiome se encuentra compuesto por 21 ASVs bacterianos y 3 fúngicos, dominados por el género Pantoea, la familia Enterobacteriacea y lo géneros fúngicos Malassezia sp. y Purpureocilium sp. Además, se encuentra un ASV asociado a la especie de controladores biológicos P. fluorescens, sugiriendo que la endósfera de plantas que no han estado expuestos a biocidas durante largos periodos de tiempo, posee gran potencial de ser reservorio de microorganismos con propiedades beneficiosas para el desarrollo vegetal y control biológico.
AGRADECIMIENTOS
Al laboratorio de Biotecnología Agrícola y de Alimentos de la Universidad San Francisco de Quito USFQ por el apoyo y financiamiento para realizar la investigación. Al Departamento de Biología de la Universidad de North Carolina, Chapel Hill por el apoyo en el secuenciamiento de las muestras. A los propietarios de las plantaciones en El Oro por permitirnos realizar el muestreo y recolectar información y a Esteban Espinosa por el apoyo en la toma de muestras.
Esta investigación se ejecutó en el Contrato Marco de Acceso a Recursos Genéticos Código: MAE-DNB-CM-2018-0085, bajo la siguiente denominación: “ESTUDIO DE DIVERSIDAD DE MICROORGANISMOS DE IMPORTANCIA MÉDICA, VETERINARIA, INDUSTRIAL, AMBIENTAL Y AGROPECUARIA DEL ECUADOR”.
CONTRIBUCIONES DE LOS AUTORES
Alejandra Paladines: realizó la investigación, el muestreo, el trabajo de laboratorio, el análisis bioinformático, la redacción del manuscrito.
Darío Ramírez: supervisión en el desarrollo de la investigación, muestreo, trabajo de laboratorio, análisis bioinformático, redacción del manuscrito.
Claudia Zapata: apoyo en la investigación, en el procesamiento de las muestras y muestreo.
Antonio León: supervisión general del proyecto y revisión del manuscrito.
CONFLICTO DE INTERESES
Los autores declaran que no existen conflictos de intereses en el presente trabajo.
REFERENCIAS
[1] Trivedi, P, Leach, J. E., Tringe, S. G., Sa, T. y Singh, B. K. (2020). Plant-microbiome interactions: from community assembly to plant health. Nature Reviews Microbiology, 18(11), 607-621. doi: https://doi.org/10.1038/s41579-020-0412-1
[2] Liu, H., Brettell, L. E. y Singh, B. (2020). Linking the Phyllosphere Microbiome to Plant Health. Trends in Plant Science, 25(9), 841-844. doi: https://doi.org/10.1016/j.tplants.2020.06.003
[3] Vorholt, J. A. (2012). Microbial life in the phyllosphere. Nature Reviews. Microbiology, 10(12), 828-840. doi: https://doi.org/10.1038/nrmicro2910
[4] Marchesi, J. R. y Ravel, J. (2015). The vocabulary of microbiome research: a proposal. Microbiome, 31(3). doi: https://doi.org/10.1186/s40168-015-0094-5
[5] Kim, M., Singh, D., Lai-Hoe, A., Go, R., Rahim, R. A., Ainuddin, A. N., Chun, J. y Adams, J. M. (2012). Distinctive Phyllosphere Bacterial Communities in Tropical Trees. Microbial Ecology, 63(3), 674-681. doi: https://doi.org/10.1007/s00248-011-9953-1
[6] Humphrey, P. T. y Whiteman, N. K. (2020). Insect herbivory reshapes a native leaf microbiome. Nature Ecology and Evolution, 4(2), 221-229. doi: https://doi.org/10.1038/s41559-019-1085-x
[7] Liu, H., Carvalhais, L. C., Crawford, M., Singh, E., Dennis, P. G., Pieterse, C. M. J. y Schenk, P. M. (2017). Inner plant values: Diversity, colonization and benefits from endophytic bacteria. Frontiers in Microbiology, 8(DEC), 1-17. doi: https://doi.org/10.3389/fmicb.2017.02552
[8] Bodenhausen, N., Somerville, V., Desiro, A., Walser, J. C., Borghi, L., Van Der Heijden, M. G. A. y Schlaeppi, K. (2019). Petunia- And Arabidopsis-specific root microbiota responses to phosphate supplementation. Phytobiomes Journal, 3(2), 112-124. doi: https://doi.org/10.1094/PBIOMES-12-18-0057-R
[9] Castrillo, G., Teixeira, P. J. P. L., Paredes, S. H., Law, T. F., De Lorenzo, L., Feltcher, M. E., Finkel, O. M., Breakfield, N. W., Mieczkowski, P., Jones, C. D., Paz-Ares, J. y Dangl, J. L. (2017). Root microbiota drive direct integration of phosphate stress and immunity. Nature, 543(7646), 513-518. doi: https://doi.org/10.1038/nature21417
[10] Lu, T., Ke, M., Lavoie, M., Jin, Y., Fan, X., Zhang, Z., Fu, Z., Sun, L., Gillings, M., Peñuelas, J., Qian, H. y Zhu, Y. G. (2018). Rhizosphere microorganisms can influence the timing of plant flowering. Microbiome, 6(1), 1-12. doi: https://doi.org/10.1186/s40168-018-0615-0
[11] Cha, J. Y., Han, S., Hong, H. J., Cho, H., Kim, D., Kwon, Y., Kwon, S. K., Crusemann, M., Bok Lee, Y., Kim, J. F., Giaever, G., Nislow, C., Moore, B. S., Thomashow, L. S., Weller, D. M. y Kwak, Y. S. (2016). Microbial and biochemical basis of a Fusarium wilt-suppressive soil. ISME Journal, 10(1), 119-129. doi: https://doi.org/10.1038/ismej.2015.95
[12] Rastogi, G., Coaker, G. L. y Leveau, J. H. J. (2013). New insights into the structure and function of phyllosphere microbiota through high-throughput molecular approaches. FEMS Microbiology Letters, 348(1), 1-10. doi: https://doi.org/10.1111/1574-6968.12225
[13] Fitzpatrick, C. R., Mustafa, Z. y Viliunas, J. (2019). Soil microbes alter plant fitness under competition and drought. Journal of Evolutionary Biology, 32(5), 438-450. doi: https://doi.org/10.1111/jeb.13426
[14] Eida, A. A., Ziegler, M., Lafi, F. F., Michell, C. T., Voolstra, C. R., Hirt, H. y Saad, M. M. (2018). Desert plant bacteria reveal host influence and beneficial plant growth properties. PLoS ONE, 13(12), 1-20. https://doi.org/10.1371/journal.pone.0208223
[15] Lei, L. (2020). Phyllosphere dysbiosis. Nature Plants, 6(5), 434. doi: https://doi.org/10.1038/s41477-020-0674-7
[16] Chen, T., Nomura, K., Wang, X., Sohrabi, R., Xu, J., Yao, L., Paasch, B. C., Ma, L., Kremer, J., Cheng, Y., Zhang, L., Wang, N., Wang, E., Xin, X. F. y He, S. Y. (2020). A plant genetic network for preventing dysbiosis in the phyllosphere. Nature, 580(7805), 653-657. doi: https://doi.org/10.1038/s41586-020-2185-0
[17] Purahong, W., Orru, L., Donati, I., Perpetuini, G., Cellini, A., Lamontanara, A., Michelotti, V., Tacconi, G. y Spinelli, F. (2018). Plant microbiome and its link to plant health: Host species, organs and pseudomonas syringae pv. Actinidiae infection shaping bacterial phyllosphere communities of kiwifruit plants. Frontiers in Plant Science, 871(November), 1-16. doi: https://doi.org/10.3389/fpls.2018.01563
[18] Smets, W. y Koskella, B. (2020). Microbiome: Insect Herbivory Drives Plant Phyllosphere Dysbiosis. Current Biology, 30 (9), R412-R414. https://doi.org/10.1016/j.cub.2020.03.039
[19] Li, P, Xu, J., Wang, Z. y Li, H. (2020). Phyllosphere Microbiome in Response to Citrus Melanose. 1-26. doi: https://doi.org/10.21203/rs.3.rs-51076/v1
[20] Evans, E. y Ballen, F. (2018). Banana Market. University of Florida. IFAS Extension, 1-9. http://edis.ifas.ufl.edu/pdffiles/FE/FE90100.pdf
[21] Churchill, A. C. L. (2011). Mycosphaerella fijiensis, the black leaf streak pathogen of banana: progress towards understanding pathogen biology and detection, disease development, and the challenges of control. Molecular Plant Pathology, 12(4), 307-328. doi: https://doi.org/10.1111/j.1364-3703.2010.00672.x
[22] Manzo-Sánchez, G., Orozco-Santos, M., Islas-Flores, I., Martínez-Bolaños, L., Guzmán-González, S., Leopardi-Verde, C. L. y Canto-Canché, B. (2019). Genetic variability of Pseudocercospora fijiensis, the black Sigatoka pathogen of banana (Musa spp.) in Mexico. Plant Pathology, 68(3), 513-522. doi: https://doi.org/10.1111/ppa.12965
[23] Kimunye, J. N., Muzhinji, N., Mostert, D., Viljoen, A., van der Merwe, A. E. y Mahuku, G. (2020). Genetic Diversity and Mating Type Distribution of Pseudocercospora fijiensis on Banana in Uganda and Tanzania. Phytopathology®. doi: https://doi.org/10.1094/PHYTO-04-20-0138-R
[24] Lu-Irving, P, Harencár, J. G., Sounart, H., Welles, S. R., Swope, S. M., Baltrus, D. A. y Dlugosch, K. M. (2019). Native and Invading Yellow Starthistle (Centaurea solstitialis) Microbiomes Differ in Composition and Diversity of Bacteria. MSphere, 4(2). doi: https://doi.org/10.1128/mSphere.00088-19
[25] Jiao, J.-Y., Wang, H.-X., Zeng, Y. y Shen, Y.-M. (2006). Enrichment for microbes living in association with plant tissues. Journal of Applied Microbiology, 100(4), 830-837. doi: https://doi.org/10.1111/j.1365-2672.2006.02830.x
[26] Finkel, O. M., Salas-González, I., Castrillo, G., Spaepen, S., Law, T. F., Teixeira, P. J. P. L., Jones, C. D., y Dangl, J. L. (2019). The effects of soil phosphorus content on plant microbiota are driven by the plant phosphate starvation response. PLOS Biology, 17(11), 1-34. https://doi.org/10.1371/journal.pbio.3000534
[27] Yourstone, S.M., Lundberg, D.S., Dangl, J.L. y Jones, C.D. (2014). MT-Toolbox: improved amplicon sequencing using molecule tags. BMC Bioinformatics, 15. doi: https://doi.org/10.1186/1471-2105-15-284
[28] Joshi, N. y Sickle, F. (2011). No Title. A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33). https://github.com/najoshi/sickle
[29] Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A. y Holmes, S. P. (2016). DADA2: High- resolution sample inference from Illumina amplicon data. Nature Methods, 13(7), 581-583. doi: https://doi.org/10.1038/nmeth.3869
[30] Prodan, A., Tremroli, V., Brolin, H., Zwinderman, A., Nieuwdrop. M. y Levin, E. (2020). Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 15(1): e0227434. doi: https://doi.org/10.1371/journal.pone.0227434
[31] Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., Robinson, C. J., Sahl, J. W., Stres, B., Thallinger, G. G., Van Horn, D. J. y Weber, C. F. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75(23), 7537-7541. doi: https://doi.org/10.1128/AEM.01541-09
[32] Gu, Z. (2020). ComplexHeatmap Complete Reference. ComplexHeatmap Complete Reference. https://jokergoo.github. io/ComplexHeatmap-reference/book/
[33] Kolde, R. (2019). pheatmap: Pretty Heatmaps. Pheatmap: Pretty Heatmaps. https://cran.r-project.org/web/packages/pheatmap/index.html
[34] Adhikari, A., Nandi, S., Bhattacharya, I., Roy, M. De, Mandal, T. y Dutta, S. (2015). Phylogenetic analysis based evolutionary study of 16S rRNA in known Pseudomonas sp. Bioinformation, 11(10), 474-480. doi: https://doi.org/10.6026/97320630011474
[35] Perazzolli, M., Antonielli, L., Storari, M., Puopolo, G., Pancher, M., Giovannini, O., Pindo, M. y Pertot, I. (2014). Resilience of the natural phyllosphere microbiota of the grapevine to chemical and biological pesticides. Applied and Environmental Microbiology, 80(12), 3585-3596. doi: https://doi.org/10.1128/AEM.00415-14
[36] Gdanetz, K. y Trail, F. (2017). The wheat microbiome under four management strategies, and potential for endophytes in disease protection. Phytobiomes Journal, 1 (3), 158-168. doi: https://doi.org/10.1094/PBI0MES-05-17-0023-R
[37] Wagner, M. R., Busby, P. E. y Balint-Kurti, P. (2020). Analysis of leaf microbiome composition of near-isogenic maize lines differing in broad-spectrum disease resistance. New Phytologist, 225(5), 2152-2165. doi: https://doi.org/10.1111/nph.16284
[38] Griffiths, S. M., Galambao, M., Rowntree, J., Goodhead, I., Hall, J., O’Brien, D., Atkinson, N. y Antwis, R. E. (2020). Complex associations between cross-kingdom microbial endophytes and host genotype in ash dieback disease dynamics. Journal of Ecology, 108(1), 291-309. doi: https://doi.org/10.1111/1365-2745.13302
[39] Luo, L., Zhang, Z., Wang, P., Han, Y., Jin, D., Su, P., Tan, X., Zhang, D., Muhammad-Rizwan, H., Lu, X. y Liu, Y. (2019). Variations in phyllosphere microbial community along with the development of angular leaf-spot of cucumber. AMB Express, 9(1). doi: https://doi.org/10.1186/s13568-019-0800-y
[40] Zhang, Z., Kong, X., Jin, D., Yu, H., Zhu, X., Su, X., Wang, P., Zhang, R., Jia, M. y Deng, Y. (2019). Euonymus japonicus phyllosphere microbiome is significantly changed by powdery mildew. Archives of Microbiology, 201(8), 1099-1109. https://doi.org/10.1007/s00203-019-01683-3
[41] Zhang, Z., Luo, L., Tan, X., Kong, X., Yang, J., Wang, D., Zhang, D., Jin, D., y Liu, Y. (2018). Pumpkin powdery mildew disease severity influences the fungal diversity of the phyllosphere. PeerJ, 2018(4), 1-16. doi: https://doi.org/10.7717/peerj.4559
[42] Hesse, C., Schulz, F., Bull, C. T., Shaffer, B. T., Yan, Q., Shapiro, N., Hassan, K. A., Varghese, N., Elbourne, L. D. H., Paulsen, I. T., Kyrpides, N., Woyke, T. y Loper, J. E. (2018). Genome-based evolutionary history of Pseudomonas spp. Environmental Microbiology, 20(6), 2142-2159. doi: https://doi.org/10.1111/1462-2920.14130
[43] Katagiri, F., Thilmony, R. y He, S. Y. (2002). The Arabidopsis thaliana-pseudomonas syringae interaction. The Arabidopsis Book, 1, e0039. doi: https://doi.org/10.1199/tab.0039
[44] Selvaraj, S., Ganeshamoorthi, P., Anand, T., Raguchander, T., Seenivasan, N., & Samiyappan, R. (2014). Evaluation of a liquid formulation of Pseudomonas fluorescens against Fusarium oxysporum f. sp. cubense and Helicotylenchus multicinctus in banana plantation. BioControl, 59, 345-355. doi: https://doi.org/10.1007/s10526-014-9569-8
[45] Bubici, G., Kaushal, M., Prigigallo, M. I., Gómez-Lama Cabanás, C. y Mercado-Blanco, J. (2019). Biological Control Agents Against Fusarium Wilt of Banana. Frontiers in Microbiology, 10, 616. doi: https://doi.org/10.3389/fmicb.2019.00616
[46] Akila, R., Rajendran, L., Harish, S., Saveetha, K., Raguchander, T. y Samiyappan, R. (2011). Combined application of botanical formulations and biocontrol agents for the management of Fusarium oxysporum f. sp. cubense (Foc) causing Fusarium wilt in banana. Biological Control, 57(3), 175-183. doi: https://doi.org/10.1016/j.biocontrol.2011.02.010