

Capacidad antioxidante del 3,4-dihidroxibenzaldehído frente al radical hidroperoxilo en disolución: Un enfoque teórico
Antioxidant capacity of 3,4-dihydroxybenzaldehyde and the hydroperoxyl radical in solution: A theoretical approach
ACI Avances en Ciencias e Ingenierías
Universidad San Francisco de Quito, Ecuador
Recepción: 28 Marzo 2019
Aprobación: 25 Septiembre 2019
Resumen: Esta investigación estudió la capacidad antioxidante del 3,4-dihidroxibenzaldehído, frente al radical hidroperoxilo por medio de un análisis termoquímico y cinético: se utilizó la Teoría de los Funcionales de la Densidad. Las energías de las barreras de las reacciones se calcularon recurriendo a la Teoría del Estado de Transición, con el nivel de teoría M05-2X/6-311+G(d,p). Los resultados termoquímicos muestran que el mecanismo de transferencia de hidrógeno es la vía principal que toma la reacción. Los cálculos cinéticos evidencian que el 3,4-dihidroxibenzaldehído es efectivo para inhibir a este radical, ya que la constante de velocidad global resultó mayor a la constante de oxidación de los ácidos grasos presentes en las membranas celulares.
Palabras clave: Cinética Química, estrés oxidativo, radical libre, capacidad antioxidante.
Abstract: Herein, we studied the antioxidant capacity of the 3,4-dihydroxybenzaldehyde against the hydroperoxyl radical by means of a thermochemical and kinetic analysis using the Theory of Functional Density. The energies of the reaction barriers were computed at the M05-2X/6-311 G(d,p) level of theory employing the Transition State Theory. The thermochemical analysis identified the hydrogen transfer mechanism as the main route taken by the reaction. The kinetic analysis indicates that 3,4-dihydroxybenzaldehyde is effective to inhibit this radical since the overall kinetic constant was higher than the oxidation constant of fatty acids present in cell membranes.
Keywords: Chemical Kinetics, oxidative stress, free radical, antioxidant capacity.
INTRODUCCIÓN
La Cinética Química se encarga del estudio de la velocidad con la que ocurren las reacciones químicas y posee una relación intrínseca con el mecanismo que las genera. Los modelos cinéticos permiten la comprensión de procesos químicos importantes como la descomposición de los alimentos, el crecimiento de microorganismos, la disgregación del ozono estratosférico o, en este caso, la determinación de la capacidad antioxidante de un compuesto [1, 2]. Desde el punto de vista teórico, los cálculos cinéticos se realizan al conocer las funciones de partición para las sustancias que participan en la reacción; entonces es posible calcular la constante de velocidad, si se emplea la teoría del estado de transición (TST, por sus siglas en inglés) [3, 4]. El método para determinar la capacidad antioxidante de un compuesto consiste en comparar la constante de velocidad del proceso, con la velocidad de oxidación de los lípidos presentes en las membranas celulares. La oxidación lipídica se genera por las especies reactivas de oxígeno durante el estrés oxidativo.
1. Estrés oxidativo
El estrés oxidativo (OS) es un proceso biológico que se desarrolla en disolución y ocurre en los organismos vivos como consecuencia del metabolismo aerobio. El OS da lugar a un entorno que se produce por el desequilibrio entre la producción de especies reactivas de oxígeno (ROS) y la capacidad del sistema biológico de reparar el daño inducido por estas especies. Parte de los ROS se generan por procesos fisiológicos que se derivan del oxígeno molecular [5], el cual produce especies como el radical anión superóxido (O2·), el radical hidroxilo (HO·), el oxígeno singulete (1O2), radicales alcoxilo (RO·), radicales peroxilo (ROO·) y, además, genera especies reactivas de nitrógeno [6]. En las células, la principal fuente de producción de ROS es la cadena respiratoria mitocondrial que utiliza aproximadamente el 80-90% del O2 que consume una persona [7]. La producción de ROS también se da por la exposición a la radiación ionizante, radiación UV o metabolismo de algunos fármacos [8].
Los ROS regulan varios procesos celulares en los mamíferos, como la producción de hormonasdecrecimiento, la comunicación entrecélulas, la unióndelas proteínas G, entre otros [9]. Cuando el nivel de especies reactivas de oxígeno supera significativamente la capacidad de las defensas antioxidantes en el sistema biológico, estas tienen efectos tóxicos en el medio [10]. La sobreproducción de ROS contribuye a la patogénesis de numerosas enfermedades neurodegenerativas relacionadas con la edad y el deterioro cognitivo en los seres humanos, debido a su ataque en los ácidos grasos insaturados de las membranas celulares y las cadenas de ADN [11].
1.1. Reactividad de las especies de oxígeno
La reactividad de los ROS se evidencia por medio del potencial de reducción; se conoce que cuanto mayor es el potencial de reducción de los ROS, mayor es su capacidad de oxidación. El peróxido de hidrógeno no se involucra directamente en el inicio de la oxidación de los lípidos, debido a que su potencial de reducción de 320 mV es inferior a los 600 mV de los ácidos grasos insaturados. El potencial de reducción del radical anión superóxido es de 940 mV: no es lo suficientemente fuerte como para extraer hidrógeno de los ácidos grasos insaturados [12]. El radical hidroxilo tiene un potencial de reducción estándar más elevado (2310 mV), pero este se genera a partir del H2O2 [13]. El radical hidroperoxilo, con 1060 mV, es más reactivo que el radical anión superóxido [14] y, por ello, su presencia en la membrana mitocondrial de las células origina la peroxidación de lípidos. La tasa de extracción de H· de los ácidos grasos insaturados varía entre 1-3 × 103 M-1 s-1, lo que representa un paso crítico en el proceso de peroxidación lipídica [12].
1.1.1 Formación del radical hidroperoxilo
El radical anión superóxido se forma en la cadena de transporte de electrones de la membrana mitocondrial, a partir del oxígeno triplete. En la célula, cuando el anión superóxido formado se difunde hacia el medio acuoso celular, forma su ácido conjugado: el radical hidroperoxilo, según la reacción (1) [15].
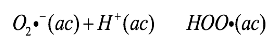 (1)
(1)Esta reacción muestra que los dos radicales se encuentran en equilibrio en medio acuoso. La reacción inversa, correspondiente a la deprotonación del HOO· tiene una constante de equilibrio, KHOO· = 1,6 x 10-5 mol/L [16]. Esto indica que, en medio acuoso, la formación del HOO· es favorable.
El radical hidroperoxilo se difunde a través de la membrana hidrofóbica [11] y produce una variedad de radicales lipídicos (R·) debido a su capacidad de extraer hidrógeno de los ácidos grasos presentes en las células, lo que da lugar a la aparición de varias enfermedades [17, 18]. Las velocidades de reacción del radical hidroperoxilo con los ácidos linoleico, linolénico y araquidónico son 1,2 × 103, 1,7 × 103 y 3,1 × 103 M-1 s-1, respectivamente [12, 19]. Estas constantes son relativamente altas y, por tanto, determinantes en los procesos oxidativos.
Algunas sustancias químicas, como los compuestos fenólicos, tienen la capacidad de inhibir el daño causado por estos radicales y ejercer un efecto protector a nivel celular [20].
2. Actividad antioxidante de los compuestos fenólicos
Los compuestos fenólicos son una clase de antioxidantes que constan de uno o más grupos hidroxilo (HO-) unidos directamente a uno o más anillos aromáticos. Los polifenoles se distribuyen ampliamente en las plantas, frutas, verduras, el vino y el té [21, 22]. Los compuestos fenólicos poseen la propiedad de prevenir, reducir o reparar el daño inducido por las especies radicales de oxígeno y nitrógeno en las biomoléculas.
El efecto inhibidor de los antioxidantes polifenólicos sobre los ROS se desarrolla por medio de mecanismos que surgen de la reacción directa con radicales libres [23]. Estos son la formación radicalaria de aductos (RAF), transferencia de hidrógeno (HT) y transferencia de electrón (SET).
En el mecanismo de transferencia de hidrógeno, los antioxidantes inactivan los radicales libres por medio de la transmisión del átomo de hidrógeno. Así, un antioxidante polifenólico genérico, ArOH, reacciona con el radical libre R· y transfiere un átomo de hidrógeno a través de la ruptura homolítica del enlace O-H, para generar radicales más estables, como se representa en la reacción (2).
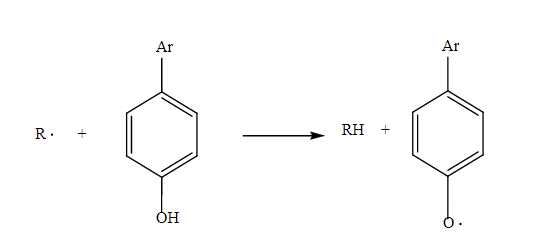 (2)
(2)Los productos de la reacción conducen a la formación de las especies RH inofensivas y el radical oxidado ArO·. El radical que se forma es menos reactivo con respecto a R· [24]. El mecanismo de transferencia de electrón permite la donación de un electrón por parte del antioxidante ArOH al radical R· según la reacción (3).
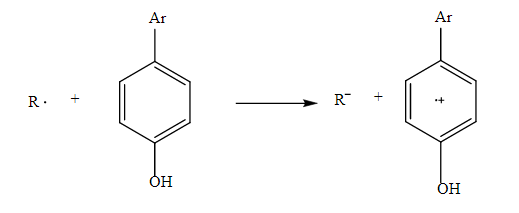 (3)
(3)El anión R- es una especie con un número par de electrones, mientras que el radical catiónico ArOH+· es una especie radical menos reactiva. En particular, el ArO· y ArOH·+ son estructuras aromáticas en las que el electrón impar -originado por las reacciones con el radical libre- tiene la posibilidad de extenderse por toda la molécula y producir una estabilización radical por resonancia [23].
Otro mecanismo que puede ocurrir entre un antioxidante y la especie radical R·, es la formación de aductos de acuerdo con la reacción (4). Esta es importante en antioxidantes que poseen dobles enlaces en su estructura, como en el caso de los carotenoides [25].
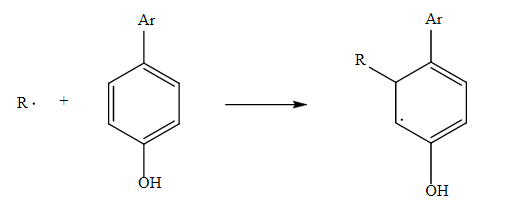 (4)
(4)El 3,4-dihidroxibenzaldehído (nombre IUPAC) o aldehído protocatéquico (PAL) es un compuesto de tipo fenólico (Fig. 1) estructuralmente similar a otras moléculas con capacidad antioxidante [26]. Estudios experimentales revelaron que este compuesto inhibe la producción de especies reactivas de oxígeno relacionadas con la aparición de la enfermedad de Parkinson [27].
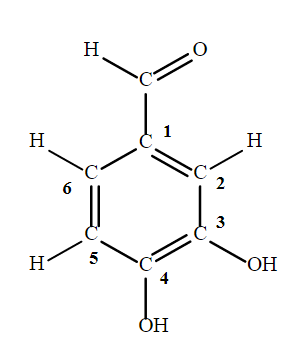
Estructura y numeración de los átomos del 3,4-dihidroxibenzaldehído. El 3,4-dihidroxibenzaldehído está presente en la salvia miltiorrhiza [28] (una de las plantas de la medicina tradicional China) y otras plantas y semillas comunes como la cebada. Jeong et al. purificaron e identificaron este compuesto en la cebada y evaluaron sus propie- dades quimiopreventivas contra el cáncer, a través de la inhibición del daño oxidativo del ADN [29].ativo del ADN [29].
2.1. Cuantificación de la actividad antioxidante
La actividad antioxidante puede expresarse como equivalentes de trolox, una unidad de medida relativa con respecto al trolox (un antioxidante de referencia) [30] o como porcentaje de radicales libres removidos o inhibidos [29]. En un estudio experimental o teórico se determina la capacidad antioxidante al saber que, si la constante de velocidad de la reacción de inhibición de radicales es mayor a la velocidad de extracción de H· de los ácidos grasos insaturados por el radical hidroperoxilo, el compuesto es efectivo para la eliminación o inhibición de estas especies [2, 31].
MATERIALES Y MÉTODOS
Los cálculos electrónicos de las especies químicas se realizaron empleando la Teoría de los Funcionales de la Densidad (DFT). En particular, se utilizó el funcional híbrido M05-2X [32], diseñado para cálculos computacionales en termoquímica y cinética [33]. Según Truhlar et al., aquel se encuentra entre los más efectivos para calcular las energías de reacción que involucran radicales libres [32]; al considerar que estos sistemas son de capa abierta, [34] se empleó el funcional correspondiente UM05-2X.
El conjunto de funciones base que se manejó es el 6-311+G(d,p) [35]: un conjunto de bases de valencia dividida con funciones de polarización (d,p) para la descripción del estado basal de cada átomo [36]. El signo + indica que la base se complementa con funciones difusas en los átomos pesados, para permitir a los orbitales ocupar un mayor espacio y describir mejor a los electrones que se encuentran distantes del núcleo. Estas funciones son básicas para representar apropiadamente a especies ionizadas y moléculas con pares de electrones no compartidos.
Para incluir los efectos del disolvente, se empleó el modelo de disolvente universal, SMD [37] que permite el cálculo de energías libres de solvatación para solutos neutros e iónicos en agua y en disolventes no acuosos. En la presente investigación, se realizaron cálculos con dos disolventes: agua y etanoato de pentilo, para imitar el medio acuoso y no polar [38, 39] de las células. El agua presenta una constante dieléctrica, ε= 78,3553 y el etanoato de pentilo ε=4,7297.
Los mínimos locales y los estados de transición se identificaron por el número de frecuencias imaginarias, 0 y 1, respectivamente. Para verificar que los estados de transición encontrados son los correctos, se efectuó el cálculo de Coordenada Intrínseca de Reacción (IRC) [40]. Este es un cálculo de trayectoria de mínima energía de reacción que permite verificar si el estado de transición se conecta con los reactivos y productos propuestos [41]. Todos estos cálculos se efectuaron con Gaussian 09 [42].
Para determinar la capacidad antioxidante por medio de la Cinética Química se empleó el programa Eyringpy, un programa escrito en Python específico para calcular propiedades termoquímicas y cinéticas [43]. Aquí se evaluaron los caminos de reacción HT, RAF y SET que tomaría el 3,4-dihidroxibenzaldehído frente al HOO·.
Las constantes de velocidad se calcularon de acuerdo con la teoría del estado de transición para los mecanismos RAF y HT. La TST es una formulación mecano-estadística para computar las constantes de velocidad de las reacciones químicas [3, 4]; se basa en el supuesto de que una reacción química avanza a lo largo de una ruta de mínima energía que conecta reactivos y productos (Fig. 2). La configuración que divide la superficie de energía potencial (PES) en regiones de reactivos y productos, se conoce como estado de transición (TS) y es un máximo en la dirección de la coordenada de reacción [44]. La TST supone que existe un quasi-equilibrio entre los reactivos y el estado de transición [45] de donde se deriva la Ec. (5),
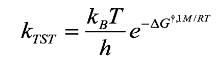 (5)
(5)en la cual kB es la constante de Boltzmann, T es la temperatura, h es la constante de Planck y R es la constante universal de los gases. Las unidades de kTST son L mol-1 s-1.
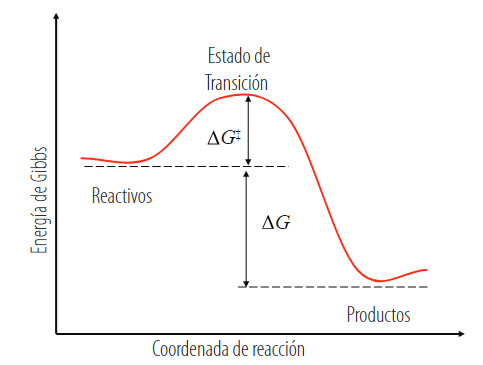
Representación unidimensional de la energía de Gibbs de una reacción.
En las reacciones que involucran transferencia de hidrógeno puede ocurrir el efecto túnel [45] Su importancia en Cinética Química radica en que la velocidad de este tipo de reacciones podría aumentar varios órdenes de magnitud [46]. El factor que permite conocer si existetunelamiento en una reacción química es el coeficientedetunelamiento o coeficiente de transmisión, el cual se introduce como un factor multiplicativo, κ, en la Ec. (5). En esta investigación, algunas de las constantes de velocidad se corrigieron por efecto túnel mediante la aproximación de Eckart [47], si κ resulta mayor que 1 existe tunelamiento y si resulta menor que 1, se lo descarta. La ecuación que incluye tal factor se representa en la Ec. (6).
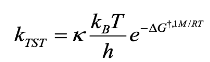 (6)
(6)En el caso de las reacciones de transferencia de electrón no ocurre la formación de un estado de transición, debido a que las moléculas de disolvente no están en equilibrio con las cargas de las especies que se oxidan y se reducen [48]. En este tipo de reacciones, las moléculas de disolvente se reorganizan, es decir se alejan o acercan de acuerdo al nuevo tamaño de las moléculas (o iones).
Marcus propuso una configuración en la que las energías de Gibbs de los reactivos y productos son iguales [48, 49]; así, la expresión matemática que permite el cálculo de la constante de velocidad para las reacciones en las que ocurre transferencia electrónica, se expresa por medio de la Ec. (7),
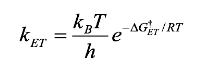 (7)
(7)donde ΔG‡ET es la energía de Gibbs de activación.
Además, las reacciones de transferencia de electrón y aquellas que involucran especies altamente reactivas, frecuentemente dependen de la difusión [31] y ocurren en procesos con barreras de activación pequeñas que hacen que la velocidad se limite por la rapidez con la que se difunden los reactivos a través del disolvente [50]; por ello, se empleó la corrección por el límite de difusión de Collins-Kimball [51] según la Ec. (8),
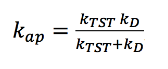 (8)
(8)donde kap es la constante de velocidad aparente; kTST es la constante de velocidad según la TST y kD es la constante de velocidad de Smoluchowski o constante de difusión; esta se calcula por medio de la Ec. (9),
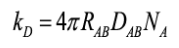 (9)
(9)donde RAB es la distancia de reacción que se obtiene a partir del TS, al medir la distancia entre los átomos que transfieren el H y la distancia entre aquellos que forman el aducto radical para las reacciones que siguen el mecanismo HT y RAF, respectivamente. Para el mecanismo SET, la distancia de reacción se obtiene a partir de la suma de los radios de los reactivos participantes. NA es el número de Avogadro y DAB es el coeficiente de difusión mutua de los reactivos A y B. DAB=DA+DB se calcula por medio de la Ec. (10) de Stokes-Einstein,
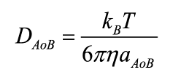 (10)
(10)donde η es la viscosidad del disolvente, en este caso η = 8,91 x 10-4 Pa.s para el agua y η = 8,62 x 10-4 Pa.s para el etanoato de pentilo; aAoB representa el radio del soluto y las unidades de DAoB y kD son m2 s-1 y L mol-1 s-1, respectivamente.
En Eyringpy se encuentran implementadas las Ec. (5) - (10) y las demás funciones de la Termodinámica Estadística, necesarias para calcular las constantes de velocidad. En el archivo de entrada de Eyringpy se ajustan el método, el efecto túnel, la difusión, temperatura, pH, distancias de reacción y disolvente, junto con los archivos de salida (out) correspondientes a los reactivos, productos y estado de transición que arroja Gaussian. Si todos los valores de entrada son correctos y sus cálculos convergen, entonces el programa arroja un archivo de salida con los correspondientes datos termoquímicos y cinéticos.
RESULTADOS
El 3,4-dihidroxibenzaldehído se representa con la notación H2C7H4O3 y sus especies ionizadas correspondientes como HC7H4O3- y C7H4O32-. El resultado de la búsqueda conformacional indica que las moléculas que forman puentes de hidrógeno intramoleculares y tienen el oxígeno del grupo aldehído en la misma orientación que los OH en el plano del anillo aromático, son estructuras de mínima energía.
Para el H2C7H4O3, el oxígeno de la posición 3 forma un puente de hidrógeno de 2,155 Å que estabiliza al sistema. Cuando el H2C7H4O3 se disocia, el hidrógeno del OH de la posición 4 se desprende para formar el HC7H4O3-, la distancia C-O se contrae 0,08 Å respecto del 3,4-dihidroxibenzaldehído sin disociar y el hidrógeno del OH de la posición 3 gira hacia el oxígeno adyacente para formar un puente de hidrógeno de 2,045 Å. En la Fig. 3 se observa que cuando el 3,4-dihidroxibenzaldehído pierde los hidrógenos, las distancias de enlace C-O de las posiciones 3 y 4 disminuyen 0,05 Å y 0,07 Å respectivamente. Para el C7H4O32- el isómero de mínima energía es aquel cuyo átomo de oxígeno del grupo aldehído tiene la misma orientación del oxígeno en la posición 3. Cada estructura de la figura hace las veces de reactivo en los diferentes mecanismos que se estudian en esta investigación. Los cambios y la reactividad de estas moléculas se indican en la Fig. 3.
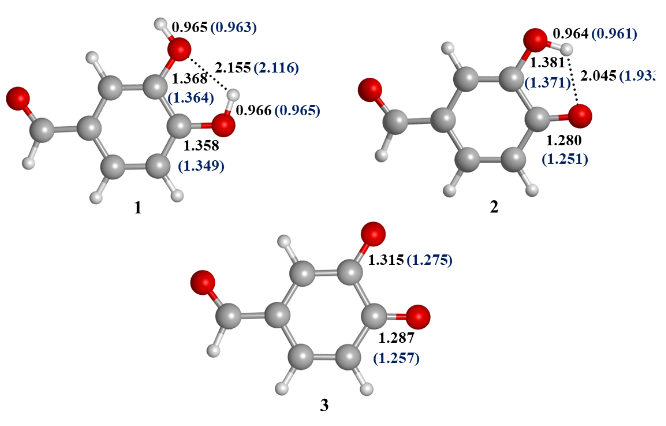
Estructura de las especies del 3,4-dihidroxibenzaldehído con el nivel M05-2X/6-311+G(d,p). Las distancias de enlace se expresan en Å. El 1 denota al H2C7H4O3, el 2 al HC7H4O3- y el 3 al C7H4O32-. Los valores entre paréntesis corresponden al medio de etanoato de pentilo.
Mecanismo de transferencia de hidrógeno
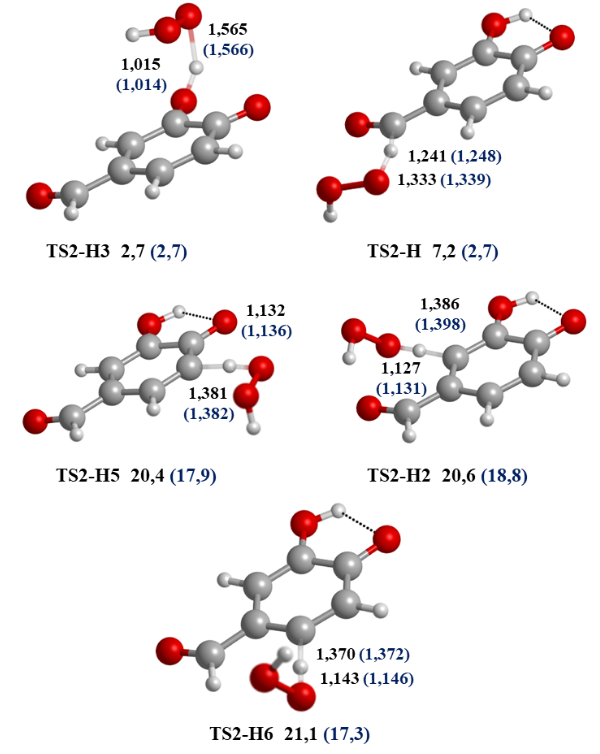
El 3,4-dihidroxibenzaldehído tiene seis hidrógenos en su estructura, los cuales pueden ser removidos por el HOO·. Las estructuras de los TS para este mecanismo se muestran en la Fig. 4. En etanoato de pentilo, los átomos de hidrógeno del 3,4-dihidroxibenzaldehído a transferirse se ubican más cerca del átomo de oxígeno del radical hidroperoxilo. Para extraer el hidrógeno del grupo aldehído y los hidrógenos 2, 5 y 6 enlazados a los carbonos, el HOO· se coloca junto al plano del anillo aromático; mientras que para extraer los hidrógenos de los grupos OH de las posiciones 3 y 4, el radical se dispone sobre el plano del anillo.
Los estados de transición correspondientes a las reacciones de transferencia del hidrógeno del grupo aldehído y los hidrógenos de los OH de las posiciones 3 y 4 son los de energía más baja. Los hidrógenos de los grupos OH son más ácidos que el H del grupo aldehído y, por tanto, su transferencia es más fácil. Ahora, entre el H3 y el H4, cuando el HOO· se aproxima, el H4 gira y se rompe el puente de hidrógeno formado con el oxígeno del OH de la posición 3 y, por ello, la transferencia del H4 cuesta 2,6 kcal/mol más que la del H3 en medio acuoso y 4,0 kcal/mol más en medio no polar.
Los hidrógenos 2, 5 y 6 enlazados a los átomos de carbono no son ácidos; su transferencia presenta barreras de energía mayor a las 20 kcal/mol y, por ello , un medio de reacción fisiológico con temperatura aproximada de 310 K no proveería tal demanda energética.
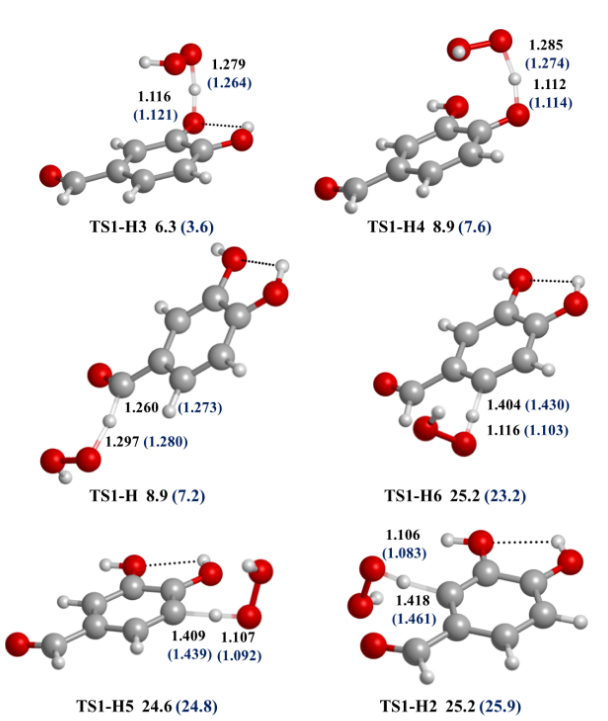
Estructura de los TS que se forman entre 1 y el HOO· en el mecanismo HT con el nivel M05-2X/6- 311+G(d,p). Las distancias en Å. En la parte inferior de cada estructura se indica el valor de la energía en kcal/mol con respecto a los reactivos. Los valores entre paréntesis corresponden al medio de etanoato de pentilo.
El parámetro termodinámico que determina la viabilidad de los mecanismos HT es el cambio de energía libre de Gibbs; si esta es menor a cero, la reacción es exergónica; si es cero, la reacción se encuentra en equilibrio, y, si es positiva, la reacción es endergónica. En la Tabla 1 se muestran los valores termoquímicos para los procesos HT entre 1 y el HOO· en ambos disolventes. La energía libre de Gibbs de los complejos de reactivos (ΔGCR) es menor que la de los reactivos. Esto ocurre porque las moléculas forman interacciones intramoleculares fuertes entre el aldehído y el radical que varían entre 7 y 9 kcal/mol y estabilizan el complejo. En cambio, la energía de Gibbs de los complejos de productos (ΔGCP) es en general mayor, debido a que el aumento de la entropía en el rompimiento de enlaces en los procesos HT, no compensa la disminución de la energía electrónica corregida con la energía de punto cero, de acuerdo con la relación ΔG=ΔH – TΔS.
| ΔGCR ΔG‡TS ΔGCP ΔGP H -9,7 (-13,6) 8,9 (7,2) -0,8 (-3,1) -7,3 (-9,3) H2 -6,0 (-9,2) 25,2 (25,7) 20,8 (18,9) 15,4 (15,7) H3 -8,3 (-9,2) 6,3 (3,6) -9,9 (-12,7) -15,0 (-17,0) H4 -6,9 (-10,6) 8,9 (7,6) -7,5 (-7,5) -12,8 (-12,0) H5 -8,3 (-10,0) 24,6 (24,8) 22,0 (22,1) 15,0 (15,5) H6 -7,1 (-10,2) 23,7 (23,2) 20,5 (21,0) 14,1 (13,9) |
Cambio de energía de Gibbs en kcal mol-1 para el mecanismo HT ente 1 y el HOO· a 298,15 K.*
* Los valores entre paréntesis corresponden al medio de etanoato de pentilo.La energía de Gibbs de reacción, calculada como la diferencia entre los reactivos y productos (ΔGP) indica que se favorecen las reacciones HT en el H del grupo aldehído y la de los hidrógenos de los OH de las posiciones 3 y 4 en ambos disolventes. El proceso más exergónico y que presenta la energía de activación más baja, es la reacción de transferencia del H3. La energía de activación efectiva (calculada desde el complejo de reactivos) es 14,6 kcal/mol para la reacción en medio acuoso. En la tabla, H hace referencia al hidrógeno del grupo aldehído; H2, H5, H6 a los hidrógenos del anillo aromático; H3 y H4 a los hidrógenos de los grupos OH en las posiciones 3 y 4.
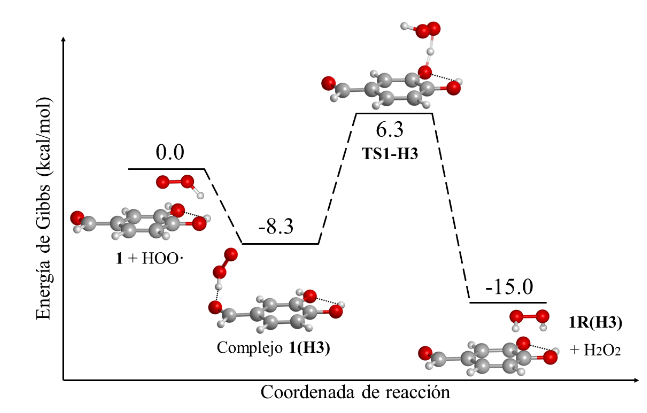
Perfil de reacción para 1 siguiendo el mecanismo HT en medio acuoso.
En la Fig. 5 se muestra el perfil de reacción de transferencia del H3 en medio acuoso. El complejo de reactivos se forma por medio de un puente de hidrógeno entre el H del radical hidroperoxilo y el O en el grupo aldehído; como productos se obtiene el radical en el oxígeno de la posición 3 más peróxido de hidrógeno. Termodinámicamente, el complejo se encuentra a -8,3 kcal/mol con respecto a los reactivos, entonces la energía de activación efectiva se calcula desde el complejo hasta el TS que se encuentra a 6,3 kcal/mol. La barrera energética real de este proceso es 14,6 kcal/mol: la más baja de todos los procesos HT, y con la reacción más negativa en la energía de Gibbs. Se espera que las reacciones que presentaron energía de Gibbs más negativa, originen productos mayoritarios. El tamaño de las barreras de energía y la formación de complejos influye en la rapidez de las reacciones, como se indica a continuación.
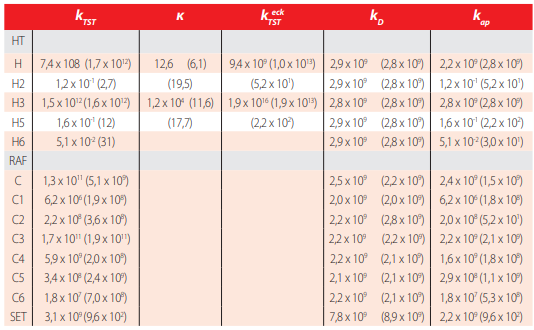
Las constantes de velocidad para las reacciones de transferencia de hidrógeno entre 1 y el HOO· se muestran en la Tabla 2. El parámetro kTST indica la constante de velocidad, sin considerar ninguna corrección. En los casos donde existe tunelamiento, el factor κ se multiplica por esta constante que da lugar a . Los valores de kap consideran la corrección por difusión y tunelamiento, en caso de existirlo.
| kTST κ kTSTeck kD kap | |||||
| H | 4,6 x 107 (8,4 x 108) | 24,0 (20,6) | 1,1 x 109 (1,7 x 1010) | 2,8 x 109 (2,9 x 109) | 7,9 x 108 (2,5 x 109) |
| H2 | 4,9 x 10-5 (1,7 x 10-5) | 2,8 x 109 (2,9 x 109) | 4,9 x 10-5 (1,7 x 10-5) | ||
| H3 | 3,7 x 109 (3,3 x 1011) | 413,1 (114,9) | 1,5 x 1012 (3,8 x 1013) | 2,6 x 109 (2,7 x 109) | 2,6 x 109 (2,7 x 109) |
| H4 | 4,8 x 107 (3,9 x 108) | 3,0 x 103 (165,1) | 1,5 x 1011 (6,4 x 1010) | 2,7 x 109 (2,7 x 109) | 2,6 x 109 (2,6 x 109) |
| H5 | 1,3 x 10-4 (1,1 x 10-4) | 2,8 x 109 (2,9 x 109) | 1,3 x 10-4 (1,1 x 10-4) | ||
| H6 | 6,2 x 10-4 (1,5 x 10-3) | 2,8 x 109 (2,9 x 109) | 6,2 x 10-4 (1,5 x 10-3) | ||
Constantes de velocidad para el mecanismo de transferencia de hidrógeno en M-1 s-1 para 1 frente al HOO· a 298,15 K.*
* Los valores entre paréntesis corresponden al medio de etanoato de pentiloEn los resultados termoquímicos se muestra que los procesos HT de los hidrógenos ácidos en medio de etanoato de pentilo presentan valores de energía de activación más baja que en medio acuoso; esto hace que el valor de kTST sea más alto en medio no polar. En ambos medios, la reacción en H3 es tan rápida que su constante tiende al límite de difusión con una rapidez de 2,6 x 109 M-1 s-1 para agua y 2,7 x 109 M-1 s-1 para etanoato de pentilo. En la reacción de transferencia del hidrógeno del OH de la posición 4, el efecto túnel que se origina por la formación de los complejos de reactivos da lugar a valores altos de κ; aumenta la rapidez de reacción y produce que la constante de velocidad tienda a la de difusión.
Las reacciones de transferencia de los hidrógenos 2, 5 y 6 presentan energías de activación elevadas; por ello, las constantes de velocidad tienden a cero y no tienen relevancia en la reactividad de ninguna de las especies del 3,4-dihidroxibenzaldehído frente al HOO·.
De igual forma, para 2 y el radical hidroperoxilo, los caminos de reacción más favorecidos son la transferencia del hidrógenodel grupo aldehído y el hidrógenodel OH dela posición 3 en ambos disolventes. La energía de activación efectiva más baja es de 10,7 kcal/mol en medio acuoso (Fig. 6) mientras que en medio apolar es de 18,9 kcal/mol: ambas para el proceso HT en el H3. La barrera para H3, en medio no polar, es significativamente más grande que en medio polar; esto explica por qué es más difícil que ocurra la reactividad de especies ionizadas en disolventes apolares.
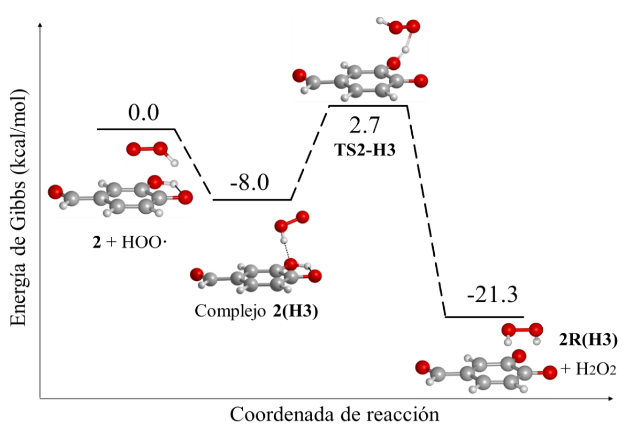
Perfil de reacción para 2 siguiendo el mecanismo HT en medio acuoso.
Para el caso de 3, la reacción viable es la transferencia del H en el grupo aldehído, la energía de activación efectiva es de 17,7 y 19,6 kcal/mol con constantes de velocidad de 2,0 x 109 y 2,8 x 109 M-1 s-1 en medio acuoso y no polar, respectivamente.
Mecanismo de formación de aductos
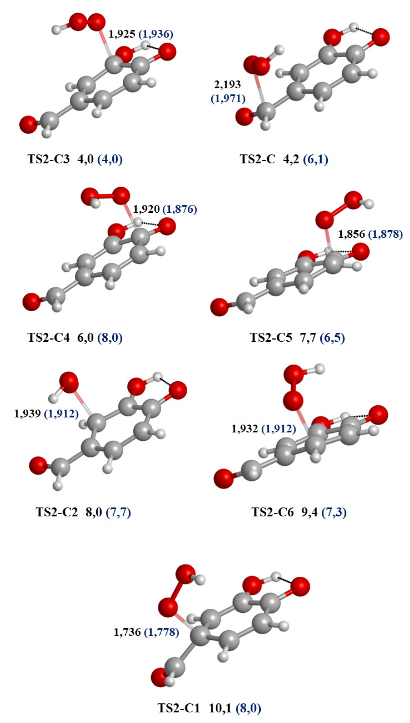
El 3,4-dihidroxibenzaldehído posee un átomo de carbono en el grupo aldehído y 6 en el anillo aromático en donde puede darse la adición del radical hidroperoxilo. La estructura de los TS se muestra en la Fig. 7. En las reacciones de formación de los aductos de los carbonos 1, 3 y 4, el radical HOO· se adiciona de tal manera que el grupo aldehído y los grupos hidroxilo de esas posiciones se inclinan un ángulo aproximado de 15°, debido a las repulsiones electrónicas entre los oxígenos.
La reacción de formación del aducto en la posición 4 presenta el estado de transición TS1-C4 como el de menor energía, le sigue el TS1-C6 correspondiente a la adición del radical en el C6. Esta tendencia indica que el mecanismo RAF se favorece en las posiciones orto y para en el anillo aromático, respecto del grupo aldehído. Según este análisis, el siguiente estado de transición sería el TS1-C2 en lugar delTS1-C3; sin embargo, la adición del radical en el C3 hace que el anillo aromático no quede totalmente plano, por lo que la distancia entre el oxígeno de la posición 4 y el hidrógeno del OH de la posición 3 aumenta a 2,195 Å, al romper el puente de hidrógeno y, por ello, existe una diferencia de 0,7 kcal/mol entre el TS1-C3 y el TS1-C2. El TS, correspondiente a la adición en el C5 y los demás procesos RAF, presenta energía más elevada y dificulta la formación de los productos en condiciones fisiológicas.
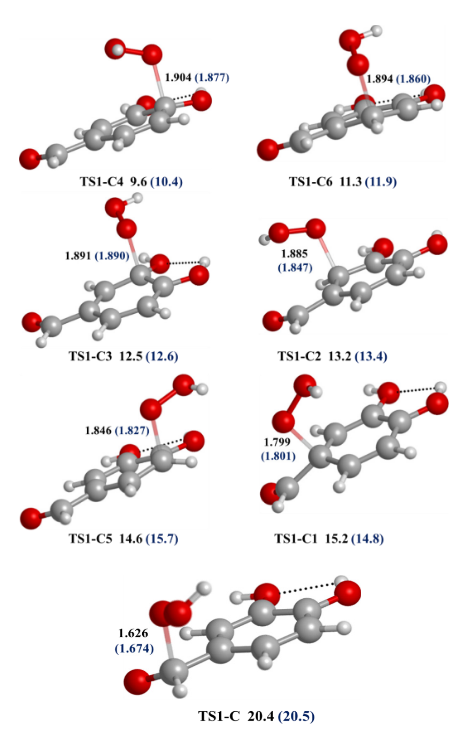
Estructura de los TS que se forman entre 1 y el HOO· en el mecanismo RAF con el nivel M05-2X/6- 311+G(d,p). Las distancias en Å. En la parte inferior de cada estructura se indica el valor de la energía en kcal/mol con respecto a los reactivos. Los valores entre paréntesis corresponden al medio de etanoato de pentilo.
De acuerdo a la Tabla 3, los complejos de reactivos se favorecen en todos los procesos de adición y los de productos -en este caso- no se forman, ya que se trata de productos de adición y no de moléculas separadas como en el caso del mecanismo HT, donde había posibilidad de formar puentes intermoleculares. El cambio de energía de Gibbs entre productos y reactivos indica que la formación del aducto en el carbono 4 (posición para) es exergónica con un ΔGP = -0,4 kcal/mol en medio acuoso. En comparación con los productos del mecanismo HT, los de adición no son mayoritarios, ya que la energía de Gibbs es ligeramente negativa y, por tanto, la constante de equilibrio (K) es pequeña según la relación ΔG = - RT Ln(K). En la tabla, el literal C hace referencia al carbono del grupo aldehído.
| ΔGCR ΔG‡TS ΔGP | |||
| C | -10,0 (-14,5) | 20,4 (20,5) | 14,7 (8,1) |
| C1 | -6,6 (-9,7) | 15,2 (14,8) | 10,8 (9,7) |
| C2 | -7,0 (-13,3) | 13,2 (13,4) | 4,6 (0,2) |
| C3 | -7,4 (-10,2) | 12,5 (12,6) | 4,0 (3,1) |
| C4 | -7,1 (-9,6) | 9,6 (10,4) | -0,4 (0,4) |
| C5 | -6,2 (-10,4) | 14,6 (15,7) | 8,5 (8,5) |
| C6 | -6,6 (-10,4) | (11,9) | 2,2 (3,0) |
Cambio de energía de Gibbs en kcal mol-1 para el mecanismo RAF entre 1 y el HOO· a 298,15 K.*
* Los valores entre paréntesis corresponden al medio de etanoato de pentiloLa Tabla 4 indica las constantes de velocidad para el mecanismo de formación de aductos para cada carbono de 1, frente al radical hidroperoxilo. En ninguno de estos procesos existe tunelamiento: el HOO· no puede atravesar las barreras de energía debido a la masa relativamente alta que posee. En general, las reacciones de formación de aductos no son espontáneas y, si ocurriesen, sus constantes de velocidad son bajas o próximas a la tasa de extracción de H en los ácidos grasos presentes en las células.
| kTST kD kap | |||
| C | 1,7 x 10-1 (1,3 x 10-1) | 2,0 x 109 (1,9 x 109) | 1,7 x 10-1 (1,3 x 10-1) |
| C1 | 1,1 x 103 (2,3 x 103) | 2,0 x 109 (2,1 x 109) | 1,1 x 103 (2,3 x 103) |
| C2 | 3,1 x 104 (2,5 x 104) | 2,1 x 109 (2,1 x 109) | 3,1 x 104 (2,5 x 104) |
| C3 | 1,0 x 105 (8,3 x 104) | 2,1 x 109 (2,2 x 109) | 1,0 x 105 (8,3 x 104) |
| C4 | 1,3 x 107 (3,4 x 106) | 2,1 x 109 (2,2 x 109) | 1,3 x 107 (3,4 x 106) |
| C5 | 2,9 x 103 (4,5 x 102) | 2,1 x 109 (2,1 x 109) | 2,9 x 103 (4,5 x 102) |
| C6 | 7,9 x 105 (3,2 x 105) | 2,1 x 109 (2,2 x 109) | 7,9 x 105 (3,2 x 105) |
Constantes de velocidad (M-1 s-1) para el mecanismo RAF de 1 frente al HOO· a 298,15 K.*
* Los valores entre paréntesis corresponden al medio de etanoato de pentiloLos procesos RAF para 2 presentan mayor número de reacciones con energía de Gibbs negativa: esto se debe a que el puente de hidrógeno que estabiliza las estructuras se mantiene en los productos. En este caso, la adición en el carbono del grupo aldehído, los carbonos 2 y 3 en medio acuoso, al igual que los carbonos 2, 3, 4 y 6 en medio no polar, son exergónicos. Cuando el disolvente es etanoato de pentilo, los complejos de reactivos presentan energía más baja en comparación con los del medio acuoso. En la Fig. 8 se muestra la reacción de formación del aducto radical en el carbono 2; esta es exergónica para ambos disolventes, con un cambio de energía de Gibbs de -0,4 en agua y -3,7 kcal/mol en medio apolar. La energía de activación efectiva es 16,2 kcal/ mol en medio acuoso y 21,3 kcal/mol en medio no polar; debido a que el complejo de reactivos en medio apolar se encuentra a aproximadamente 5,4 kcal/mol más abajo en la superficie de energía potencial. Estos datos termoquímicos explican, además, por qué las reacciones de especies ionizadas en disolventes apolares no ocurren.
Las reacciones de adición en el C7H4O32- presentan barreras de energía que tienden a 0 kcal/mol con respecto a 3 + HOO·.
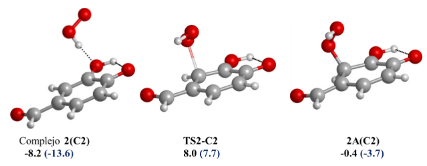
Estructura del complejo de reactivos, estado de transición y producto de adición correspondientes a la reacción de formación del aducto en C2 con el nivel M05-2X/6-311+G(d,p). En la parte inferior de cada estructura se indica el valor de la energía en kcal/mol con respecto a 2 + HOO·. Los valores entre paréntesis corresponden al medio de etanoato de pentilo.
Mecanismo de transferencia de electrones
El H2C7H4O3 no participa en el mecanismo de transferencia de electrones; estos procesos resultan demasiado endergónicos, ya que es difícil transferir electrones desde una especie neutra como el H2C7H4O3 hacia el radical. El valor de energía de Gibbs de reacción es 23,8 kcal/mol cuando el disolvente es agua y 67,4 kcal/mol en medio de etanoato de pentilo. La energía de activación que deben alcanzar estos procesos es 23,9 y 81,6 kcal/mol, en medio acuoso y no polar, respectivamente.
La termoquímica de los procesos SET refleja la dificultad que presentan las especies ionizadas para reaccionar en disolventes apolares; así, para 2 en medio acuoso la reacción (11) es exergónica con un ΔG= -2,5 kcal/mol y una energía de activación de 4,5 kcal/mol; mientras que en medio no polar, la reacción es endergónica con ∆G= 11,8 kcal/mol y una barrera de 13,4 kcal/mol.
 (11)
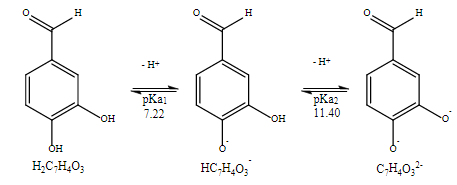
(11)En general, 3 presenta valores elevados de las constantes de transferencia de electrón que en principio hacen pensar que el C7H4O32- es la especie predominante en el sistema; sin embargo, al analizar el diagrama de distribución de especies del 3,4-dihidroxibenzaldehído (Fig.9) se observa que en condiciones fisiológicas (pH=7,4) el C7H4O32- tiene una fracción molar de 1,8 x 10-5, esto significa que la concentración de esta especie y las reacciones que genera son prácticamente despreciables.
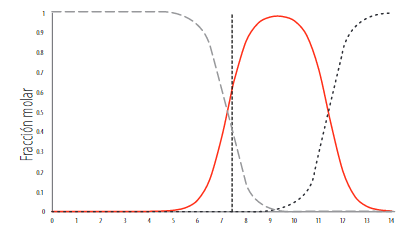
Diagrama de distribución de especies del 3,4-dihidroxibenzaldehído en función del pH.
Actividad antioxidante del 3,4-dihidroxibenzaldehído
La fracción molar del HC7H4O3- es 0,602 y la del H2C7H4O3 es 0,398, lo que indica que el monoanión es la especie que se encuentra en mayor proporción. Ahora, al considerar todos los mecanismos se deduce que el mecanismo HT es el principal camino de reacción que tomaría el 3,4-dihidroxibenzaldehído para reducir la toxicidad del radical hidroperoxilo, ya que estos procesos reportan las constantes de velocidad con los órdenes de magnitud más altos, los valores de energía de activación efectiva más bajos, así como los valores de cambio de energía libre de Gibbs más negativos.
Para el proceso en medio acuoso, en el cual el monoanión transfiere un protón al radical hidroperoxilo, se tiene una constante de velocidad k1 según la reacción (12).
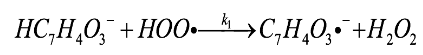 (12)
(12)En una reacción paralela, el HC7H4O3- transfiere un electrón hacia el HOO· con una constante de velocidad k2 según la reacción (13).
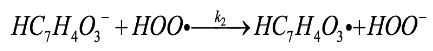 (13)
(13)Adicionalmente, la especie C7H4O3·-, producto de la transferencia de hidrógeno también cede un electrón al radical según la reacción (14).
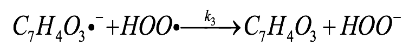 (14)
(14)Las ecuaciones diferenciales que gobiernan estos procesos se reflejan en las Ec. (15) y (16)
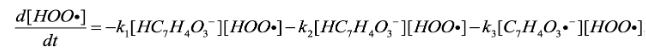 (15)
(15)
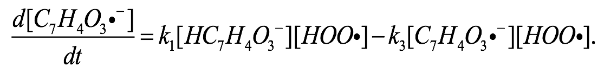 (16)
(16)Para encontrar [C7H4O3·-] se emplea la aproximación del estado estacionario en la Ec. (15):
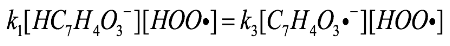 (17)
(17)entonces:
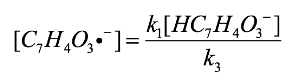 (18)
(18)al reemplazar la expresión de la Ec. (18) en la Ec. diferencial (16) se obtiene la Ec. (19):
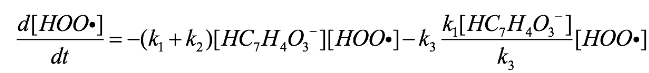 (19)
(19)y al simplificar se obtiene la Ec. (20):
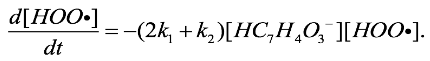 (20)
(20)Como puede notarse en la Ec. (20), la constante de velocidad global para la inhibición del radical HOO· corresponde a 2k1 + k2. El valor de k1 es 2,8 x 109 M-1 s-1 mientras que el valor de k2 es 2,2 x 109 M-1 s-1 (ver Tabla S.2 en Material Suplementario); es decir, la constante global tiene un valor de 7,8 x 109 M-1 s-1. Al constituir la Ec. (20) se obtiene la ley integrada de velocidad del proceso de acuerdo con la Ec. (21).
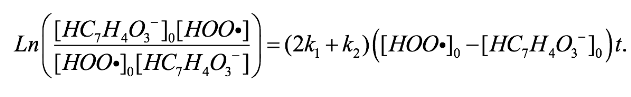 (21)
(21)donde [HOO·]0 y [HC7H4O3-]0 son las concentraciones iniciales del radical hidroperoxilo y el monoanión, respectivamente, siendo [HC7H4O3-]0 = 0,602CH2C7H4O3 (ver Material Suplementario). Esta ecuación permite conocer las concentraciones instantáneas del radical y el aldehído en función del tiempo.
En ambiente no polar, el H2C7H4O3 es la especie química que predomina al transferir un hidrógeno al radical según la reacción (22).
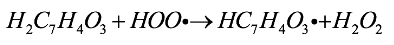 (22)
(22)Esta reacción tiene una constante de velocidad k = 2,7 x 109 M-1 s-1 ( Tabla 2); al considerar que las constantes de velocidad correspondientes al daño que causa el HOO· en los ácidos grasos poliinsaturados, están en el rango de 1-3 x 103 M-1 s-1 [12], se predice que el 3,4-dihidroxibenzaldehído tiene la capacidad de inhibir estos radicales.
DISCUSIÓN
La capacidad antioxidante del 3,4-dihidroxibenzaldehído frente al radical hidroperoxilo se estudió por medio de un análisis termoquímico y cinético en dos disolventes: agua para imitar el ambiente acuoso del citosol presente dentro de la células y etanoato de pentilo para imitar el medio no polar de la membrana celular, donde se generan estos radicales.
El análisis conformacional indica que las especies químicas más estables para el 3,4-dihi- droxibenzaldehído poseen estructuras planas con un enlace de hidrógeno intramolecular. Este resultado coincide con un análisis que se realizó con el nivel de teoría B3LYP/6-31G [52].
Termodinámicamente, se encontró que las reacciones de transferencia de los hidrógenos ácidos del compuesto fenólico presentan los cambios de energía libre de Gibbs de reacción más negativos y las barreras de energía más bajas, lo que significa que el mecanismo HT es la vía principal que toma la reacción. A medida que aumenta el grado de deprotonación del aldehído, se favorecen las reacciones de adición en los átomos de carbono de la molécula; sin embargo, la exergonicidad de estos procesos sigue siendo menor a los procesos HT.
La constante de velocidad para la reacción entre el ácido 3,4-dihidroxibenzóico y el HOO· es 1,26 x 107 M-1 s-1 en medio acuoso y 5,14 x 103 M-1 s-1 en medio de etanoato de pentilo [39]: esto sugiere que la capacidad antioxidante del 3,4-dihidroxibenzaldehído es mayor que la del ácido 3,4-dihidroxibenzóico. Los ensayos experimentales de capacidad antioxidante equivalente de trolox (TEAC) y el ensayo de 2,2-difenil-1-picrililhidracilo (DPPH·) [53] junto con los cálculos de varios descriptores moleculares en DFT realizados para estos compuestos, confirman esta tendencia [52]. Los resultados en ambiente no polar concuerdan con las propiedades de los antioxidantes fenólicos de retardar la oxidación de lípidos a través del mecanismo de transferencia de hidrógeno [54].
El mecanismo secuencial de transferencia de los hidrógenos 3 y 4 del aldehído se estudia, además, desde el punto de vista teórico; siempre y cuando se considere el carácter multirreferencial del proceso: su importancia radica en la determinación de la capacidad prooxidante del compuesto.
AGRADECIMIENTOS
El autor agradece al Consejo Nacional de Ciencia y Tecnología (CONACyT) y al HCP Kukulcán-Cinvestav Mérida, México Proyecto CB-2015-252356, por la asignación de recursos computacionales; a la M. en C. Eugenia Dzib Reyes por su ayuda en el manejo del programa Eyringpy.
Material suplementario. Este trabajo generó una gran cantidad de estructuras que comprende reactivos, complejos, estados de transición y productos. Las coordenadas de estas estructuras se encuentran en un archivo electrónico que detalla primero las de medio acuoso y, luego, las correspondientes al etanoato de pentilo. Se adjunta, además, un documento que contiene las tablas y figuras para los mecanismos de reacción de la especie ionizada del 3,4-dihidroxibenzaldehído y el cálculo de la distribución de especies en función del pH.
REFERENCIAS
[1] Gorban, A. N., & Yablonsky, G. S. (2015). Three waves of chemical dynamics. Mathematical Modelling of Natural Phenomena, 10(5), 1-5. doi: https://doi.org/10.1051/mmnp/201510501
[2] Maroz, A., Anderson, R. F., Smith, R. A., & Murphy, M. P. (2009). Reactivity of ubiquinone and ubiquinol with superoxide and the hydroperoxyl radical: implications for in vivo antioxidant activity. Free Radical Biology and Medicine, 46(1), 105-109. doi: https://doi.org/10.1016/j.freeradbiomed.2008.09.033
[3] Evans, M. G., & Polanyi, M. (1935). Some applications of the transition state method to the calculation of reaction velocities, especially in solution. Transactions of the Faraday Society, 31, 875-894.
[4] Eyring, H. (1935). The activated complex in chemical reactions. The Journal of Chemical Physics, 3(2), 107-115. doi: https://doi.org/10.1063/1.1749604
[5] Buonocore, G., Perrone, S., & Tataranno, M. L. (2010, August). Oxygen toxicity: chemistry and biology of reactive oxygen species. In Seminars in Fetal and Neonatal Medicine (Vol. 15, No. 4, pp. 186-190). WB Saunders. doi: https://doi.org/10.1016/j.siny.2010.04.003
[6] Losada-Barreiro, S., &Bravo-Diaz, C. (2017). Free radicals and polyphenols:The redox chemistry of neurodegenerative diseases. European Journal of Medicinal Chemistry, 133, 379-402. doi: https://doi.org/10.1016/j.ejmech.2017.03.061
[7] Rosini, M., Simoni, E., Milelli, A., Minarini, A., & Melchiorre, C. (2013). Oxidative Stress in Alzheimer’s Disease: Are We Connecting the Dots? Miniperspective. Journal of medicinal chemistry, 57(7), 2821-2831. doi: https://dx.doi.org/10.1021/jm400970m
[8] Lee, J., Koo, N., & Min, D. B. (2004). Reactive oxygen species, aging, and antioxidative nutraceuticals. Comprehensive reviews in food science and food safety, 3(1), 21-33. doi: https://doi.org/10.1111/j.1541-4337.2004.tb00058.x
[9] Bartosz, G. (2009). Reactive oxygen species: destroyers or messengers?. Biochemical pharmacology, 77(8), 1303- 1315. doi: https://doi.org/10.1016/j.bcp.2008.11.009
[10] Valko, M., Rhodes, C., Moncol, J., Izakovic, M. M., & Mazur, M. (2006). Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-biological interactions, 160(1), 1-40. doi: https://doi.org/10.1016/j.cbi.2005.12.009
[11] Silva, T., Reis, J., Teixeira, J., & Borges, F. (2014). Alzheimer’s disease, enzyme targets and drug discovery struggles: from natural products to drug prototypes. Ageing research reviews, 15, 116-145. doi: https://doi.org/10.1016/j.arr.2014.03.008
[12] Bielski, B. H., Arudi, R. L., & Sutherland, M. W. (1983). A study of the reactivity of HO2/O2-with unsaturated fatty acids. Journal of Biological Chemistry, 258(8), 4759-4761.
[13] Choe, E., & Min, D. B. (2005). Chemistry and reactions of reactive oxygen species in foods. Journal of food science, 70(9), R142-R159. doi: https://doi.org/10.1111/j.1365-2621.2005.tb08329.x
[14] Gus’ kova, R. A., Ivanov, I. I., Kol’tover, V. K., Akhobadze, V. V., & Rubin, A. B. (1984). Permeability of bilayer lipid membranes for superoxide (O2−) radicals. Biochimica et BiophysicaActa (BBA)-Biomembranes, 778(3), 579-585. doi: https://doi.org/10.1016/0005-2736(84)90409-7
[15] Sawyer, D.T., & Valentine, J. S. (1981). How super is superoxide?. Accounts of Chemical Research, 14(12), 393-400. doi: https://doi.org/10.1016/0005-2736(84)90409-7
[16] Bielski, B. H., Cabelli, D. E., Arudi, R. L., & Ross, A. B. (1985). Reactivity of HO2/O− 2 radicals in aqueous solution. Journal of physical and chemical reference data, 14(4), 1041-1100. doi: https://doi.org/10.1063/1.555739
[17] Brand, M. D., Affourtit, C., Esteves, T. C., Green, K., Lambert, A. J., Miwa, S., ... & Parker, N. (2004). Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radical Biology and Medicine, 37(6), 755-767. doi: https://doi.org/10.1016/j.freeradbiomed.2004.05.034
[18] Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T., Mazur, M., &Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. The international journal of biochemistry & cell biology, 39(1), 44-84. doi: https://doi.org/10.1016/j.biocel.2006.07.001
[19] Aikens, J., & Dix, T. A. (1991). Perhydroxyl radical (HOO.) initiated lipid peroxidation. The role of fatty acid hydroperoxides. Journal of Biological Chemistry, 266(23), 15091-15098.
[20] Cai,Y.,Luo,Q.,Sun,M.,&Corke,H.(2004).Antioxidantactivityandphenoliccompoundsof112traditionalChinesemedicinal plants associated with anticancer. Life sciences, 74(17), 2157-2184. doi: https://doi.org/10.1016/j.lfs.2003.09.047
[21] Scalbert, A., & Williamson, G. (2000). Dietary intake and bioavailability of polyphenols. The Journal of nutrition, 130(8), 2073S-2085S. doi: https://doi.org/10.1093/jn/130.8.2073S
[22] Stevenson, D. E., & Hurst, R. D. (2007). Polyphenolic phytochemicals–just antioxidants or much more?. Cellular and Molecular Life Sciences, 64(22), 2900-2916. doi: https://doi.org/10.1007/s00018-007-7237-1
[23] Leopoldini, M., Marino, T., Russo, N., & Toscano, M. (2004). Antioxidant properties of phenolic compounds: H-atom versus electron transfer mechanism. The Journal of Physical Chemistry A, 108(22), 4916-4922. doi: https://dx.doi.org/10.1021/jp037247d
[24] Leopoldini, M., Russo, N., & Toscano, M. (2011). The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chemistry, 125(2), 288-306. doi: https://doi.org/10.1016/j.foodchem.2010.08.012
[25] Galano, A., & Francisco-Marquez, M. (2009). Reactions of OOH radical with β-carotene, lycopene, and torulene: hydrogen atom transfer and adduct formation mechanisms. The Journal of Physical Chemistry B, 113(32), 11338- 11345. doi: https://pubs.acs.org/doi/full/10.1021/jp904061q
[26] Kakkar, S., & Bais, S. (2014). A review on protocatechuic acid and its pharmacological potential. ISRN pharmacology, 2014. doi: http://dx.doi.org/10.1155/2014/952943
[27] Gao, J. W., Yamane, T., Maita, H., Ishikawa, S., Iguchi-Ariga, S. M., Pu, X. P., & Ariga, H. (2010). DJ-1–Mediated Protective Effect of Protocatechuic Aldehyde Against Oxidative Stress in SH-SY5Y Cells. Journal of pharmacological sciences, 1012100484-1012100484. doi: https://doi.org/10.1254/jphs.10271FP
[28] Gu, M., Wang, X., Su, Z., & Ouyang, F. (2007). One-step separation and purification of 3, 4-dihydroxyphenyllactic acid, salvianolic acid B and protocatechualdehyde from Salvia miltiorrhiza Bunge by high-speed counter-current chromatography. Journal of Chromatography A, 1140(1-2), 107-111. doi: https://doi.org/10.1016/j.chroma.2006.11.057
[29] Jeong, J. B., Hong, S. C., & Jeong, H. J. (2009). 3, 4-Dihydroxybenzaldehyde purified from the barley seeds (Hordeum vulgare) inhibits oxidative DNA damage and apoptosis via its antioxidant activity. Phytomedicine, 16(1), 85-94. doi: https://doi.org/10.1016/j.phymed.2008.09.013
[30] Dávalos, A., Gómez-Cordovés, C., & Bartolomé, B. (2004). Extending applicability of the oxygen radical absorbance capacity (ORAC− fluorescein) assay. Journal of agricultural and food chemistry, 52(1), 48-54. doi: https://dx.doi.org/10.1021/jf0305231
[31] Galano, A., & Alvarez‐Idaboy, J. R. (2013). A computational methodology for accurate predictions of rate constants in solution: Application to the assessment of primary antioxidant activity. Journal of computational chemistry, 34(28), 2430-2445. doi: https://doi.org/10.1002/jcc.23409
[32] Zhao, Y., Schultz, N. E., & Truhlar, D. G. (2006). Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. Journal of Chemical Theory and Computation, 2(2), 364-382. doi: https://dx.doi.org/10.1021/ct0502763
[33] Peverati, R., & Truhlar, D. G. (2012). Performance of the M11 and M11-L density functionals for calculations of electronic excitation energies by adiabatic time-dependent density functional theory. Physical Chemistry Chemical Physics, 14(32), 11363-11370. doi: https://dx.doi.org/10.1039/c2cp41295k
[34] Szabo, A., & Ostlund, N. S. (2012). Modern quantum chemistry: introduction to advanced electronic structure theory. Courier Corporation.
[35] Binkley, J. S., Pople, J. A., & Hehre, W. J. (1980). Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. Journal of the American Chemical Society, 102(3), 939-947. doi: https://doi.org/10.1021/ja00523a008
[36] Krishnan, R. B. J. S., Binkley, J. S., Seeger, R., & Pople, J. A. (1980). Self‐consistent molecular orbital methods. XX. A basis set for correlated wave functions. The Journal of Chemical Physics, 72(1), 650-654. doi: https://doi.org/10.1063/1.438955
[37] Marenich, A. V., Cramer, C. J., & Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. The Journal of Physical Chemistry B, 113(18), 6378-6396. doi: https://dx.doi.org/10.1021/jp810292n
[38] Alberto, M. E., Russo, N., Grand, A., & Galano, A. (2013). A physicochemical examination of the free radical scavenging activity of Trolox: mechanism, kinetics and influence of the environment. Physical chemistry chemical physics, 15(13), 4642-4650. doi: https://dx.doi.org/10.1039/c3cp43319f
[39] Galano, A., & Pérez-González, A. (2012). On the free radical scavenging mechanism of protocatechuic acid, regeneration of the catechol group in aqueous solution. Theoretical Chemistry Accounts, 131(9), 1265. doi: https://dx.doi.org/10.1007/s00214-012-1265-0
[40] Fujisaki, N., Ruf, A., & Gäumann, T. (1987). Barrier permeabilities for a symmetric Eckart potential as studied by the kinetic isotope effects for hydrogen/deuterium abstraction from neopentane by hydrogen atoms in the gas phase. Journal of the Chemical Society, Faraday Transactions 2: Molecular and Chemical Physics, 83(11), 2053-2065. doi: https://dx.doi.org/10.1039/F29878302053
[41] Gonzalez, C., & Schlegel, H. B. (1989). An improved algorithm for reaction path following. The Journal of Chemical Physics, 90(4), 2154-2161. doi: https://doi.org/10.1063/1.456010
[42] Frisch, M. J., et al. (2009). Gaussian 09. Gaussian, Inc.: Wallingford CT.
[43] Dzib, E., Cabellos, J. L., Ortíz‐Chi, F., Pan, S., Galano, A., & Merino, G. (2019). Eyringpy: a program for computing rate constants in the gas phase and in solution. International Journal of Quantum Chemistry, 119(2), e25686. doi: https://doi.org/10.1002/qua.25686
[44] Henriksen, N. E., & Hansen, F. Y. (2018). Theories of molecular reaction dynamics: the microscopic foundation of chemical kinetics. Oxford University Press.
[45] Zhang, Y., Stecher, T., Cvitaš, M. T., & Althorpe, S. C. (2014). Which Is Better at Predicting Quantum-Tunneling Rates: Quantum Transition-State Theory or Free-Energy Instanton Theory?. The journal of physical chemistry letters, 5(22), 3976-3980. doi: https://dx.doi.org/10.1021/jz501889v
[46] Liu,Y. P., Lu, D. H., Gonzalez-Lafont, A., Truhlar, D. G., & Garrett, B. C. (1993). Direct dynamics calculation of the kinetic isotope effect for an organic hydrogen-transfer reaction, including corner-cutting tunneling in 21 dimensions. Journal of the American Chemical Society, 115(17), 7806-7817. doi: https://dx.doi.org/10.1021/ja00070a029
[47] Eckart, C. (1930). The penetration of a potential barrier by electrons. Physical Review, 35(11), 1303. doi: https://doi.org/10.1103/PhysRev.35.1303
[48] Marcus, R. A. (1993). Electron transfer reactions in chemistry. Theory and experiment. Reviews of Modern Physics, 65(3), 599. doi: https://doi.org/10.1103/RevModPhys.65.599
[49] Marcus, R. A. (1956). On the theory of oxidation‐reduction reactions involving electron transfer. I. The Journal of Chemical Physics, 24(5), 966-978. doi: https://doi.org/10.1063/1.1742723
[50] Truhlar, D. G. (1985). Nearly encounter-controlled reactions: The equivalence of the steady-state and diffusional viewpoints. Journal of Chemical Education, 62(2), 104. doi: https://dx.doi.org/10.1021/ed062p104
[51] Collins, F. C., & Kimball, G. E. (1949). Diffusion-controlled reaction rates. Journal of colloid science, 4(4), 425-437. doi: https://doi.org/10.1016/0095-8522(49)90023-9
[52] Nenadis, N., & Tsimidou, M. Z. (2012). Contribution of DFT computed molecular descriptors in the study of radical scavenging activity trend of natural hydroxybenzaldehydes and corresponding acids. Food research international, 48(2), 538-543. doi: https://doi.org/10.1016/j.foodres.2012.05.014
[53] Bountagkidou, O. G., Ordoudi, S. A., & Tsimidou, M. Z. (2010). Structure–antioxidant activity relationship study of natural hydroxybenzaldehydes using in vitro assays. Food research international, 43(8), 2014-2019. doi: https://doi.org/10.1016/j.foodres.2010.05.021
[54] Frankel, E. N. (1998). Lipid Oxidation. Dundee. Scotland: The Oily Press LTD.
MATERIAL SUPLEMENTARIO
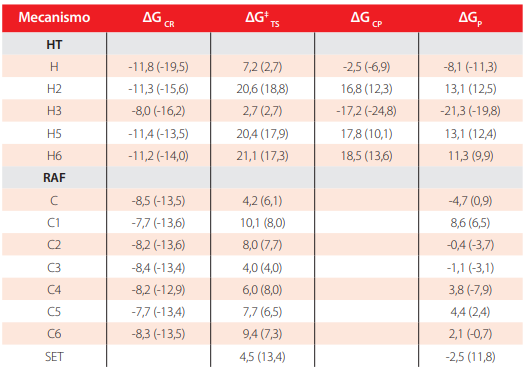
Cambio de energía de Gibbs en kcal mol-1 para 2 frente al HOO· a 298,15 K.*
* Los valores entre paréntesis corresponden al medio de etanoato de pentilo.
Constantes de velocidad (M-1 s-1) para 2 frente al HOO· a 298,15 K.*
* Los valores entre paréntesis corresponden al medio de etanoato de pentilo.
Estructura de los TS que se forman entre 2 y el HOO· en el mecanismo HT con el nivel M05-2X/6- 311+G(d,p). Las distancias en Å. En la parte inferior de cada estructura se indica el valor de la energía en kcal/mol con respecto a los reactivos. Los valores entre paréntesis corresponden al medio de etanoato de pentilo.
Estructura de los TS que se forman entre 2 y el HOO· en el mecanismo RAF con el nivel M05-2X/6- 311+G(d,p). Las distancias en Å. En la parte inferior de cada estructura se indica el valor de la energía en kcal/mol con respecto a los reactivos. Los valores entre paréntesis corresponden al medio de etanoato de pentilo.
Cálculo de la fracción molar para las especies del 3,4-Dihidroxibenzaldehído en función del pH.
En medio acuoso, la fracción molar de las especies H2C7H4O3, HC7H4O3- y C7H4O32- se calculan por medio de los valores de pKa según el esquema de la Figura S.3.
Disociación del 3,4-dihidroxibenzaldehído en medio acuoso.
La concentración de las especies de H2C7H4O3, HC7H4O3- y C7H4O32- dependen, en este caso del valor del pH fisiológico (7.4) y se representan por medio de las reacciones de equilibrio (S.1) y (S.2).
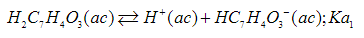 (S.1)
(S.1)
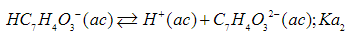 (S.2)
(S.2)La constante de equilibrio para ka1 se representa en la Ec. (S.3).
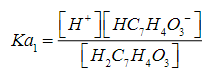 (S.3)
(S.3)el balance de masa da lugar a la Ec. (S.4).
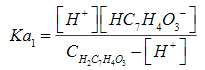 (S.4)
(S.4)resolviendo para las Ec. (S.3) y (S.4) se obtiene la Ec. (S.5).
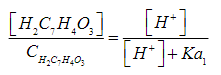 (S.5)
(S.5)donde el cociente [H2C7H4O3]/CH2C7H4O3 representa la fracción molar para el H2C7H4O3 (XH2C7H4O3), usando las relaciones: [H+]=10-pH y Ka=10-pKa para el valor del pH fisiológico se tiene que XH2C7H4O3 = 0,398. De igual forma, para el HC7H4O3- se tiene obtiene la Ec. (S.6).
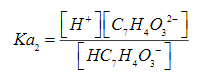 (S.6)
(S.6)
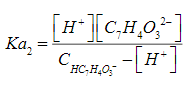 (S.7)
(S.7)de las relaciones anteriores se tiene que [ HC7H4O3- ] = CH2C7H4O3 – [ H2C7H4O3 ] y CH2C7H4O3- ≈ [ HC7H4O3- ] , entonces resolviendo para las Ec. (S.6) y (S.7) se obtiene la Ec. (S.8).
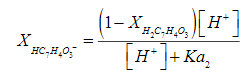 (S.8)
(S.8)análogamente se obtiene que XHC7H4O3-=0,602. Para encontrar H2C7H4O3, HC7H4O3- y XC7H4O32- se sabe que la suma de las fracciones molares es igual a uno, de este modo se obtiene que XC7H4O32- es 1,8 x 10 -5.