

Distribución del 1-butanol y 2-butanol en los sistemas agua/n-octano y agua/Dodecil Sulfato de Sodio (SDS)/n-octano usando dinámica molecular. Parte II. Uso de las herramientas gmx-density y gmx-densmap
Distribution of the 1-butanol and 2-butanol in the water/ n-octane and water/Sodium Dodecyl Sulfate (SDS)/n-octane systems using molecular dynamics. Importar imagen Importar imagen Importar imagenPart II. Use of the gmx-density and gmx-densmap tools
ACI Avances en Ciencias e Ingenierías
Universidad San Francisco de Quito, Ecuador
Recepción: 13 Noviembre 2018
Aprobación: 08 Enero 2019
Resumen: En este trabajo, la distribución de las moléculas de 1-butanol y 2-butanol en los sistemas agua/n-octano y agua/SDS/n-octano fue determinada usando las herramientas gmx- density y gmx- densmap del programa gromacs con la finalidad de complementar a nivel computacional el comportamiento experimental estos co-surfactantes cuando están localizados en la región in- terfacial de estos sistemas. Los modelos de energía potencial GROMOS53A6 y SPC fueron utilizados para describir a las moléculas de 1-butanol, 2-butanol, SDS y agua, respectivamente. Estos modelos fueron capaces de predecir las propiedades interfaciales del sistema agua/n-octano y el área por molécula del Dodecil Sulfato de Sodio en la interfaz agua/n-octano de forma consistente. Finalmente, los perfiles y mapas de densidad demuestran que las moléculas de alcohol y SDS coexisten en la región interfacial del sistema agua/n-octano favoreciendo la estabilidad de la monocapa de surfactante y la película interfacial.
Palabras clave: Dinámica molecular, perfiles de densidad, región interfacial, surfactantes, co-surfactantes.
Abstract: In this paper, the distribution of 1-butanol and 2-butanol molecules in the water/n- octane and water/SDS/n-octane systems was determined using the gmx-density and gmx-densmap tools with the aim of explore the behavior of these co-surfactants when are located at the interfacial region of these systems. The GROMOS53A6 and SPC force fields were used to describe to the 1-butanol, 2-butanol, SDS and water molecules, respectively. These force fields predict the interfacial properties of the water/n-octane and area per molecule of the Dodecyl Sulfate of So- dium of form consistent. Finally, density profiles and density maps demonstrate that the alcohol and SDS molecules coexist in the interfacial region of the water/n-octane system favoring the stability of the surfactant monolayer and the interfacial film.
Keywords: Molecular dynamic, density profile, interfacial region, surfactants, co-surfactants.
INTRODUCCIÓN
Los surfactantes y co-surfactantes son especies que contienen grupos hidrofóbicos e hidrofíli- cos [1,2]. Estas moléculas pueden modificar las propiedades de ciertas superficies y regiones interfaciales [3,4]. El comportamiento de los surfactantes y co- surfactantes en las regiones interfaciales aire/agua y agua/aceite han sido estudiadas por sus aplicaciones industriales en el área de lubricación, emulsiones, química ambiental y atmosférica, recuperación mejorada de crudos y formación de películas delgadas [5-9].
Diferentes estudios experimentales han demostrado que los surfactantes adoptan una posición preferencial en la región interfacial de los sistemas aire/agua y agua/aceite, la cual es significativamente diferente a la distribución aleatoria e isotrópica de los surfactantes en el seno de un líquido como el agua [10-13]. Técnicas experimentales como rayos X, reflactancia de neutrones y espectroscopía vibracional de suma de frecuencias han permitido determinar la organización estructural de monocapas de surfactantes localizados en la región interfacial de líquidos in- miscibles, aportando información a nivel molecular sobre el espesor de las monocapas, de la distribución de contraiones y de la orientación de los solventes alrededor de los surfactantes [14-17].
De igual manera, se ha estudiado el efecto de la carga del grupo hidrofílico del surfactante, las características del grupo lipofílico, la interacción entre el aceite y la cadena hidrocarbonada del surfactante, y la mezcla de surfactantes en la monocapa. Estas investigaciones han permitido demostrar que los sistemas formados por surfactantes y co-surfactantes muestran un comportamiento más favorable que permite aumentar la rigidez y estabilidad de la película interfacial utilizando una menor cantidad de surfactante [18-21].
El comportamiento de las mezclas de Dodecil Sulfato de Sodio (SDS) con alcoholes han sido estudiadas experimentalmente en diferentes sistemas aire/agua y agua/aceite [2]. El SDS es un surfactante altamente hidrofílico que se caracteriza por tener una buena capacidad para reducir la tensión interfacial de los sistemas aire/agua y agua/aceite,aumentando esta capacidad con la presencia de co-surfactantes [22,23].
Adicionalmente, se han propuesto diferentes ecuaciones semiempíricas donde se relacionan las diferencias de los potenciales químicos del surfactante en agua y en aceite con los diferentes parámetros de formulación como la concentración de surfactante, la concentración de alcohol y la temperatura del sistema [24,25].
De igual manera, los alcoholes de cadena hidrocarbonada corta (3 carbonos o menos) como propanol y los alcoholes de cadena hidrocarbonada larga (5 carbonos o más) como pentanol han sido usados ampliamente como co-surfactantes. Estos alcoholes tienen la capacidad de ad- sorberse en la region interfacial de los sistemas agua/aceite mejorando la actividad interfacial de la mezcla anfifílica. Los alcoholes de cadena corta como el propanol aumentan ligeramente la hidrofilicidad de la mezcla surfactante/alcohol en la región interfacial, mientras que los alcoholes de cadena larga como pentanol aumentan considerablemente la lipofilicidad de la mezcla surfactante/alcohol [26].
En cambio, el comportamiento de los alcoholes de cadena intermedia como el 1-butanol y 2-butanol depende de la naturaleza del surfactante utilizado y de las variables de fomulación como la salinidad y el tipo de hidrocarburo. Por ejemplo, cuando el 1-butanol es más lipofílico que el surfactante, un incremento de la concentración de este co-surfactante, ocasiona un aumento de la lipofilicidad de la mezcla anfifílica en la region interfacial [27].
De hecho, la distribución de los componentes de un sistema SDS/1-butanol/agua/aceite ha si- do determinada a diferentes relaciones de mezcla agua/aceite usando el diagrama de fase tipo α – β – ε [22]. En este trabajo, Chai et al. demostraron que la presencia de surfactante en la región interfacial genera una disminución de la concentración de 1-butanol. También, la afinidad de este alcohol en la región interfacial aumenta en función de la longitud de la cadena hidrocarbonada del aceite utilizado. A su vez, se ha sugerido que los alcoholes coexisten con las moléculas de surfactantes localizadas en la monocapa interfacial reduciendo su interacción molecular y produciendo una mezcla de surfactante/alcohol desordenada en la región interfacial [28].
A nivel molecular, la dinámica molecular ha sido empleada para estudiar el comportamiento interfacial de surfactantes en sistemas aire/agua y agua/aceite donde se forman monocapas, bicapas y micelas [29-33]. De hecho, en un trabajo previo, Parra y Aray determinaron el comportamiento interfacial del SDS localizado en la región interfacial agua/n-octano [34]. En dicho trabajo, se logró determinar el espesor de la película interfacial y la tensión interfacial de los sistemas agua/n-octano, vacío/SDS/n-octano y agua/SDS/n-octano. Adicionalmente, en este tra- bajo, se determinó el área máxima que ocupa una molécula de SDS en la región interfacial del sistema agua/n-octano y el arreglo molecular de la monocapa de dicho surfactante en la región interfacial.
También, esta técnica de simulación ha sido empleada para determinar las propiedades inter- faciales de las mezclas surfactante/surfactante y surfactante/alcohol de cadena larga en los sis- temas aire/agua y agua/aceite[35-38]. Sin embargo, la distribución de alcoholes de cadena intermedia como 1-butanol y 2-butanol en el sistema agua/SDS/n- octano no ha sido reportado a nivel molecular.
Por tal motivo, en este trabajo, se evaluó el alcance de las herramientas gmx-density y gmx- densmap del programa GROMACS para determinar la distribución de 1-butanol y 2-butanol en los sistemas agua/n-octano y agua/SDS/n-octano, particulamente en la región interfacial. Esto se hizo con la finalidad de complementar a nivel computacional el comportamiento experimental de estos co-surfactantes en estos sistemas. Para ello, los modelos de energía potencial GRO- MOS53A6[39] y SPC[40] se utilizaron para describir las moléculas de 1-butanol, 2-butanol, SDS, n-octano y agua, respectivamente. Un total de seis sistemas fueron simulados con dinámica molecular y la metodología emplea- da fue propuesta por J. Parra et al. (datos sin publicar) en un trabajo previo que se encuen- tra en proceso de revisión donde se estimó la tensión interfacial, los perfiles de densidad y la función de distribución radial para determinar las interacciones moleculares del surfactante y co-surfactante en la región interfacial.
A diferencia del trabajo de J. Parra et al. (datos sin publicar) que se encuentra en proceso de revisión, el análisis propuesto en este manuscrito fue realizado desde una perspectiva estructural en la región interfacial. En este caso se determinó la distribución de los alcoholes en la región interfacial en presencia de SDS por inspección visual de la configuración final y se corroboró la distribución de los alcoholes usando las herramientas gmx-density y gmx-densmap.
MATERIALES Y MÉTODOS
Fundamentos teóricos y detalles computacionales
El modelo de energía potencial
En la dinámica molecular, el modelo de energía potencial describe a nivel molecular las espe- cies involucradas en una simulación. Aquí, la energía total de un sistema de moléculas, Etotal, es determinada como la suma de las contribuciones de las interacciones intramoleculares e in- termoleculares. En este caso, la energía total es descrita por la ecuación 1,
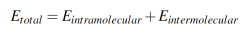 (1)
(1)donde Eintramoleculares la energía intramolecular asociada a los enlaces, ángulos entre enlaces y ángulos dihedrales formados entre cuatro átomos. En cambio, el término Eintermolecularcorresponde a la suma de las interacciones de van der Waals y coulómbicas entre los átomos de las especies presentes en el sistema [41].
En este trabajo, El modelo GROMOS53A6[39] describe las interacciones moleculares entre 1-butanol, 2-butanol y SDS. Los parámetros enlazantes y las cargas atómicas de las molécu- las fueron obtenidos del servidor Automated force field Topology Builder (ATB) [42] con una geometría optimizada a un nivel de cálculo B3LYP/6-31G*.
En las topologías de las moléculas, la distribución de cargas que propone ATB no fue utiliza- da. En este trabajo, dichas distribuciones de cargas fueron construídas usando el procedimiento propuesto por J. Parra et al. (datos sin publicar), formando grupos moleculares con dos, tres, cuatro y cinco átomos (los grupos moleculares corresponden a grupos metilenos, metilos, oxi-drilos, sulfato e iones). A su vez, el modelo GROMOS53A6 calcula la energía no enlazante, Ene, mediante la ecuación 2,
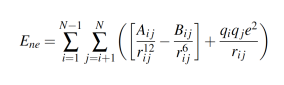 (2)
(2)Donde, Aij, Bij, qi, q j and rijson los parámetros de la energía de interacción no enlazante, las cargas atómicas y las distancias interatómicas entre las partículas i y j, respectivamente. Las estructuras moleculares de 1-butanol, 2-butanol y SDS son mostradas en la figura 1.
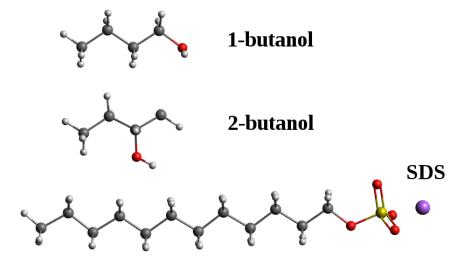
Estructuras moleculares del 1-butanol, 2-butanol y Dodecil Sulfato de Sodio (SDS).
Finalmente, las moléculas de agua fueron descritas con el modelo de punto de carga simple (Simple Point Charge, SPC) [40]. En la topología del agua, la distancia O-H es igual a 0.100 nm y el ángulo H-O-H fue de 109.47°. En el modelo SPC existe una única interacción de Lennard- Jones entre los átomos de oxígeno. Los parámetros de Lennard- Jones y las cargas atómicas en el modelo SPC son σ (O) = 0.317 nm, ε(O) = 0.650 kJ/mol, q(H) = 0.4100 e y q(O) =-0.8200 e. Este modelo ha permitido predecir las propiedades macroscópicas del agua como la densidad.
Propiedades interfaciales
El espesor de película interfacial y la tensión interfacial son propiedades que se presentan en un sistema formado por dos líquidos inmiscibles.
En un sistema líquido/líquido, la región interfacial comienza, donde la densidad de ambos líquidos disminuye con respecto al seno del líquido puro. Para determinar la región interfacial y el espesor de película interfacial en un modelo de bicapas es necesario graficar los perfiles de densidad. El perfil de densidad de una especie tipo i en la dirección perpendicular a la región interfacial se describe por la ecuación 3:
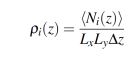 (3)
(3)Donde, ρi(z) es el perfil de densidad a lo largo del eje z, (Ni(z)) es el número de especies tipo i entre z – z/2 y z + z/2 en el tiempo t [35]. El intervalo entre capas, ∆z ,fue igual anm 0.02 nm.
La tensión interfacial se determina con el modelo mecánico de Kirwood-Buff[43] usando los tensores de presión como se muestra en la ecuación 4,
 (4)
(4)Donde, PN y PT son los componentes normal y tangencial de la presión, respectivamente. Lz es la dimensión de la celda de simulación a lo largo del eje z. El componente normal PN es igual a Pzz, mientras el componente tangencial PT es dado por 1/2(Pxx + Pyy). Lostensores de presión Pxx, Pyy y Pzz son definidos de forma general mediante la ecuación 5,
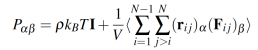 (5)
(5)Donde I es el tensor unitario, kB es la constante de Boltzmann, T es la temperatura, y p = N/V es la densidad en número. Los términos . y . representan las direcciones X, Y, y Z. En la ecuación 5, ri jrepresenta el vector entre el centro de masa de la molécula i y j. El término Fi j es la fuerza intermolecular entre moléculas i y j, la cual se expresa como la suma de todas las fuerzas interactuantes [44].
La tensión interfacial se determina a lo largo del eje z con los tensores de presión promedio mediante la ecuación 6,
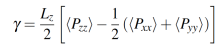 (6)
(6)Construcción de los sistemas simulados
El procedimiento utilizado en este trabajo fue el propuesto por J. Parra et al. (datos sin publicar), cuyo manuscrito se encuentra en proceso de revisión.
Con el programa Avogadro 1.0[45] se construyeron las moléculas de 1-butanol, 2-butanol, agua y SDS. Las coordenadaas xyz de las configuraciones obtenidas fueron almacenadas en el for- mato Protein Data Bank (PDB).
Los seis sistemas simulados (agua/n-octano, agua/1-butanol/n-octano, agua/2- butanol/n-octano, agua/SDS/n-octano, agua/SDS+1-butanol/n-octano y agua/SDS+2- butanol/n-octano), se cons- truyeron con las herramientas insert-molecules y editconf del programa GROMACS versión 5.0.2 [46-48]. Un total de 360 moléculas de n-octano y 1600 moléculas de agua fueron colo- cadas en las celdas periódicas rectangulares de dimensiones 4x4x3 nm3. Las densidades de los sistemas n-octano y agua iniciales fueron803 Kg/m3 y 997 Kg/m3, respectivamente. Luego, con estas celdas se construyeron lascapas de agua y n-octano utilizadas en la preparación del sistema agua/n-octano. Lascapas de n-octano fueron colocadas en ambos lados de la capa de agua para producirdos regiones interfaciales en el sistema agua/n-octano.
Seguidamente, las monocapas de 1-butanol, 2-butanol y SDS fueron construídas con un total de 25 moléculas y cubriendo un área total de 4x4 nm2. Estas monocapas fueron colocadas en la región interfacial agua/n-octano para formar los sistemas agua/1butanol/n-octano,agua/2-butanol/n-octano yagua/SDS/n-octano.
Con este mismo procedimiento se construyeron los sistemas agua/SDS+1-butanol/n- octano y agua/SDS+2-butanol/n-octano. Un total de 25 moléculas de alcohol con 25 moléculas de SDS fueron colocadas en la región interfacial del sistema agua/n-octano generando una relación de mezcla igual a 1:1.
Condiciones de las simulaciones realizadas
Todas las simulaciones fueron realizadas con el programa GROMACS versión 5.0.2 [46-48]. Las configuraciones iniciales se optimizaron con el método steepest descent para obtener los sistemas con mínima energía.
Los sistemas agua/n-octano y agua/SDS/n-octano fueron equlibrados con una dinámica molecular tipo NPT de 20 ns a 300 K y 1 bar. Seguidamente, a estos sistemas se les realizó una segunda simulación tipo NPT de 20 ns bajo las mismas condiciones de presión y temperatura para determinar las propiedades interfaciales. Este procedimiento fue utilizado con la finalidad de evaluar la capacidad de los modelos GROMOS53A6 y SPC para predecir las propiedades interfaciales.
Luego, los sistemas construídos fueron tratados con una simulación tipo NP.T de 500 ps a una temperatura de 300 K y con una presión normal al plano xy de 1050 bar. Este procedimiento de simulación permite comprimir los sistemas a lo largo del eje z manteniendo constante el área por molécula en el plano xy. A continuación, a las configuraciones finales obtenidas, se les realizó una simulación tipo NPT de 20 ns a las mismas condiciones de temperatura y presión (300 K y 1 bar) para determinar las propiedades interfaciales y las distribución de las moléculas de surfactantes y co- surfactantes en los sistemas. En este caso, este procedimiento fue usado para evaluar la distribución de los alcoholes en el sistema agua/n-octano en presencia de SDS. En las simulaciones de dinámica molecular, la temperatura fue controlada usando el termostato modificado de Berendsen (velocity-rescaling thermostat en inglés)[49] y la presión con el ba- rostato de berendsen[50] con una constante de acoplamiento igual a 0.1 ps. Las distancias de enlace fueron restringidas usando el algoritmo LINCS[51] y las condiciones periódicas de la celda fueron aplicadas en todas las direcciones. El algoritmo de salto de rana[52] fue utilizado para la integración de las ecuaciones de Newton con un paso del tiempo de 1 fs y las coordenadas almacenadas cada 1 ps. Las interacciones de van der Waals fueron determinadas con un radio de interacción de 1.4 nm y las interacciones electrostáticas estimadas con el método de Ewald[53]. Las gráficas fueron realizadas con el programa GRACE[54] y las imagénes de los sistemas moleculares con el programa VMD [55]. En todas las simulaciones de dinámica molecular tipo NPT, la energía potencial, la densidad de los sistemas y el cubrimiento superficial (área por molécula) de los surfactantes y co-surfactantes se mantuvo constante a lo largo de la trayectoria.
RESULTADOS
Capacidad de los modelos GROMOS53A6 y SPC para predecir las propiedades de los sistemas agua/n-octano y agua/SDS/n-octano
Los modelos de energía potencial GROMOS53A6 y SPC fueron usados en conjunto para estudiar el comportamiento de los sistemas agua/n-octano y agua/SDS/n-octano y evaluar su capacidad para predecir las propiedades interfaciales de los sistemas mencionados. Para ello, las configuraciones finales en equilibrio fueron extráıdas de las simulaciones de dinámica molecu- lar tipo NPT para su análisis (ver figura 2(a) y 2(b)).
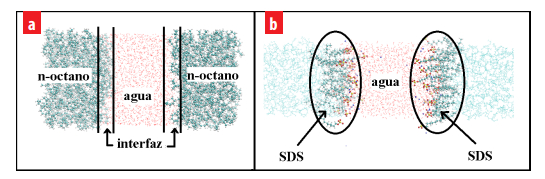
Configuraciones finales de los sistemas (a) agua/n-octano y (b) agua/SDS/n-octano, obtenidas usando dinámica molecular tipo NPT. En estos sistemas, las moléculas de agua están en el centro del sistema y el n-octano a los extremos. Las moléculas de SDS están localizadas en la región interfacial.
La figura 2(a) representa la configuración en equilibrio del sistema agua/n-octano. Claramente, se puede observar la presencia de dos regiones interfaciales bien definidas en el sistema. En dichas regiones interfaciales, las moléculas de agua y n-octano interaccionan entre sí. Aquí, las moléculas de n-octano localizadas en la zona interfacial se ubican normal al plano interfacial para minimizar la repulsión con las moléculas de agua.
La densidad del sistema agua/n-octano fue de 869.48 ± 0.17 Kg/m3. Esta densidad se encuentra comprendida entre las densidades experimentales del agua y del n-octano (997 Kg/m3 y 718 Kg/m3, respectivamente). Esta densidad es consistente con respecto a los valores experimentales.
En la figura 2(b), se puede observar como las moléculas de SDS estan localizadas exclusivamente en la región interfacial del sistema agua/n-octano. El grupo sulfato (–OSO−3 ) del SDS penetra la capa de agua debido a su naturaleza hidrofílica para obtener una buena solvatación y las cadenas hidrocarbonadas se muestran extendidas y ligeramente inclinadas en la capa de n-octano. En cambio, los iones Sodio ( Na+) están distribuidos en la capa de agua y cercanos a la región interfacial.
Este fenómeno es consistente con el reportado en otras simulaciones y experimentos, donde el surfactante se distribuye uniformemente en la región interfacial formando una monocapa rígida que permite la variación de la tensión interfacial del sistema agua/n- octano [21,29-31,34].
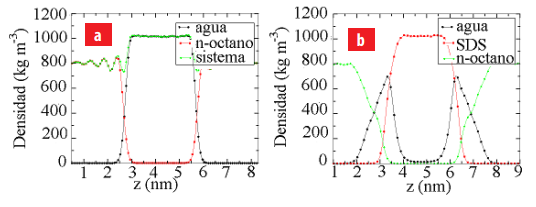
Distribución de la densidad de los diferentes componentes a lo largo del eje z. (a) Perfil de densidad del sistema agua/n-octano y (b) Perfil de densidad del sistema agua/SDS/n- octano.
En la figura 3(a) y 3(b), se muestran los perfiles dedensidad delos diferentes componentes que conforman los sistemas agua/n-octano y agua/SDS/n-octano a lo largo del eje z.
Las densidades promedios obtenidas de la simulación en el seno de los líquidos agua y n-octano fueron 803.18 Kg/m3 and 1003.10 Kg/m3 (ver figura 3(a)). De igual manera, estos resultados están en concordancia con los valores reportados experimentalmente para el agua y el n-octano. En la figura 3(a), la distribución de los perfiles de densidad del agua y n-octano corrobora la presencia de dos regiones interfaciales localizados en el eje z. En la zona interfacial, el perfil de densidad del n-octano presenta ciertas fluctuaciones cerca de la capa de agua que desaparecen
cuando se alejan de la zona interfacial. La presencia de estos picos corresponden a la posición del mínimo de energía del potencial de lennard-Jones para los átomos de carbono. Este mismo comportamiento fue observado en otros trabajos [56,57]. Estas fluctuaciones están asociadas al arreglo espacial que deben tener las moléculas de n-octano en la región interfacial para reducir las interacciones repulsivas con las moléculas de agua. A su vez, el perfil de densidad del agua es uniforme a lo largo del eje z.
En la figura 3(b), se muestran los perfiles de densidad de los componentes presentes en el sistema agua/SDS/n-octano. El SDS se encuentra localizado en la región interfacial del sistema agua/n-octano. El perfil de densidad del SDS esta ubicado en la región interfacial, indicando que los grupos –OSO−3 y los iones Na+ coexisten con el agua y la cadenahidrocarbonada del SDS interacciona con el n-octano. De igual manera, la presencia dela monocapa de SDS genera un ensanchamiento de la región interfacial agua/n-octanoaumentando la cantidad de moléculas de agua y n-octano en esta zona.
A su vez, el cubrimiento superficial de la región interfacial del sistema agua/n-octano por el SDS fue determinada de las simulaciones de dinámica molecular tipo NPT y la tensión interfacial de los sistemas agua/n-octano y agua/SDS/n-octano fue estimada con el modelo de Kirkwood y Buff [43].
La tensión interfacial obtenida para el sistema agua/n-octano fue igual a 54.89 ± 0.71 mN/m. Este valor se encuentra en excelente concordancia con los reportados experimentalmente de 51.68 mN/m y 52.50 mN/m [58,59].
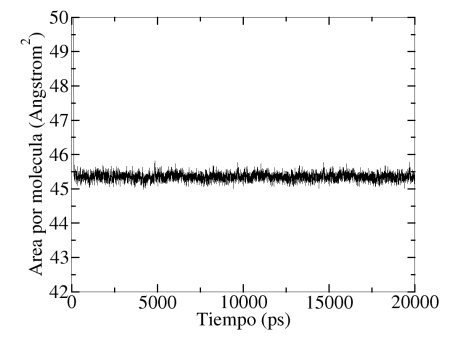
Área/molécula del SDS en función del tiempo de simulación. La monocapa de SDS cubre toda la región interfacial agua/n-octano.
En la figura 4, se muestra la variación del cubrimiento superficial (área/molécula) de la región interfacial por parte del SDS en función del tiempo de simulación.
El área/molécula fue invariante después de cierto tiempo de simulación. Este comportamiento indica que la región interfacial del sistema agua/n-octano esta completamente saturada por la monocapa de SDS.
Con la modificación realizada a la distribución de cargas de los grupos moleculares, los valores de área/molécula y tensión interfacial fueron iguales a 45.45 ± 0.30 Å 2/molécula y 2.29 ± 0.56 mN/m, respectivamente. En un trabajo previo, Parra y Aray reportaron un área/molécula de 45.60 Å 2/molécula usando la distribución de moléculas de ATB sin modificaciones y la tensión interfacial para este cubrimiento superficial fue de 4.94 ± 0.30 mN/m [34]. Estos valores son comparables con los reportados experimentalmentepor Penfold[60] de 45 Å 2/molécula para la saturación de la región interfacial aire/aguapor parte del SDS y por Rehfeld[61] de 8.35 mN/m para la tensión interfacial de unsistema agua/n-octano saturado con SDS. Aquí, pode- mos mencionar que el modelode energía potencial GROMOS53A6 sobreestima la capacidad de reducción de latensión interfacial del surfactante SDS. En este caso, sería adecuado hacer un ajuste delos parámetros no enlazantes del modelo de energía potencial para obtener con mayorexactitud la tensión interfacial del sistema agua/SDS/n-octano .
Sin embargo, todos estos resultados demuestran que la combinación de los modelos GRO- MOS53A6 y SPC son capaces de describir cualitativamente y cuantitativamente los sistemas agua/n-octano y agua/SDS/n-octano. También, estos modelos son capaces de evaluar el com- portamiento y predecir la tendencia de las propiedades interfaciales de estos sistemas.
Distribución del 1-butanol y 2-butanol en los sistemas agua/1-butanol/n- octano, agua/2- butanol/n-octano, agua/SDS+1-butanol/n-octano y agua/ SDS+2-butanol/n-octano. Uso de la herramienta gmx-density
La localización final de las moléculas de 1-butanol y 2-butanol en los sistemas agua/1- butanol/n-octano, agua/2-butanol/n-octano, agua/SDS+1-butanol y agua/SDS+2- butanol/n-octano después de las simulaciones con dinámica molecular tipo NPT son mostradas en la figura 5.
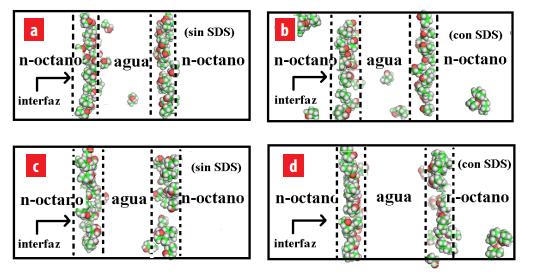
Distribución de los alcoholes en los sistemas agua/n-octano y agua/SDS/n-octano. (a) 1-butanol en agua/n-octano, (b) 1-butanol en agua/SDS/n-octano, (c) 2-butanol en agua/n- octano y (d) 2-butanol en agua/ SDS/n-octano. Las moléculas de agua, n-octano y SDS no son mostradas para obtener una mejor visualización.
En el sistema agua/n-octano, el 1-butanol presenta una mayor afinidad por la región interfacial (ver figura 5(a)). Sin embargo, debido a la solubilidad de este alcohol en agua, algunas molécu- las de 1-butanol están localizadas dentro de la capa de agua. De hecho, el grupo polar de las moléculas de 1-butanol se encuentran orientado hacia el seno de la fase acuosa cuando están localizadas en la región interfacial.
En el sistema agua/SDS/n-octano, las moléculas de 1-butanol se distribuyen en la región inter- facial y en las capas de agua y n-octano (ver figura 5(b)). La presencia de 1-butanol en el agua hace esta capa menos polar. En cambio, su presencia en la capa de n-octano la hace más polar. Este fenómeno favorece el balance hidrofílico-lipofílico del sistema agua/ SDS/n-octano. Este comportamiento fue sugerido por Salager et al. en sus estudios experimentales [24].
Seguidamente, la ubicación final de las moléculas de 2-butanol en los sistemas agua/n-octano y agua/SDS/n-octano se muestran en las figuras 5(c) y 5(d). Las moléculas de 2-butanol prefieren estar ubicadas en la región interfacial del sistema agua/n-octano cuando no existen moléculas de SDS en el sistema (ver figura 5(c)). En cambio, el 2-butanol se encuentra ubicado tanto en la región interfacial como en la capa de n-octano en el sistema agua/SDS/n-octano. Debido a la naturaleza hidrofóbica del 2-butanol, no existe la presencia de moléculas de 2-butanol en el seno de la fase acuosa.
Para verificar lo observado en las configuraciones finales, los perfiles de densidad de los sis- temas agua/1-butanol/n-octano, agua/2-butanol/n-octano, agua/SDS+1-butanol y agua/SDS+2- butanol/n-octano fueron determinadas a lo largo del eje z. Estos perfiles de densidad son mos- trados en la figura 6.
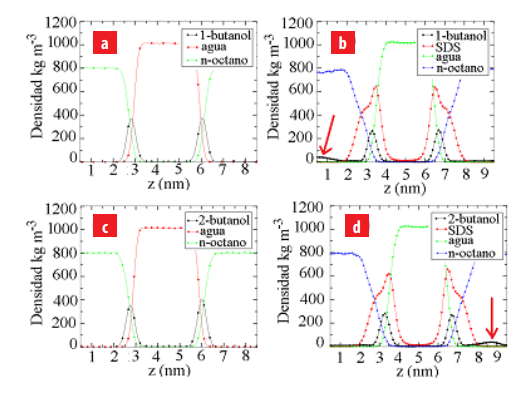
Perfiles de densidad de los componentes presentes en los sistemas estudiados. (a) agua/1-bu- tanol/n-octano, (b) agua/SDS+1-butanol/n-octano, (c) agua/2-butanol/n-octano y (d) agua/SDS+2-buta-nol/n-octano. Las flechas en rojo indican el incremento del perfil de densidad de los alcoholes en la fase n-octano debido a la presencia de SDS en la interfaz.
En la figura 6(a), se puede observar como las moléculas de 1-butanol se concentran princi- palmente en la capa de agua y en la región interfacial del sistema agua/n-octano. Este perfil de densidad del 1-butanol presenta un máximo de densidad localizados en el medio de la re- gión interfacial y se hacen cero en la fase n-octano. Sin embargo, cuando la monocapa de SDS está presente en la región interfacial del sistema agua/n-octano, las moléculas de 1-butanol mi- gran a la capa de n-octano generando un ligero incremento en su perfil de densidad en esta fase (ver figura 6(b)). Esto indica que cuando una monocapa de SDS está localizada en la región in- terfacial del sistema agua/n-octano, las moléculas de 1-butanol están presentes en las diferentes fases del sistema (capa de agua, capa de n-octano y región interfacial) modificando la naturaleza polar y no polar de las fases.
De igual manera, el 2-butanol prefiere estar en la región interfacial del sistema agua/n- octano interaccionando con las moléculas de agua y n-octano presentes en esta región (ver figura 6(c)). Sin embargo, con la presencia de la monocapa de SDS en la región interfacial del sistema agua/n-octano, el perfil de densidad en la figura 6d muestra que las moléculas de 2-butanol migran preferencialmente a la fase n-octano generando la aparición de un pico en el perfil de densidad de esta especie en la fase n-octano. Esta migración es debida a la naturaleza hidrofóbi- ca de las moléculas de 2-butanol y su mayor afinidad por el n-octano.
Adicionalmente, los perfiles de densidad de los alcoholes y SDS coexisten en la región interfa- cial del sistema agua/n-octano, lo cual demuestra que las moléculas de 1-butanol y 2-butanol en la región interfacial se distribuyen dentro de la monocapa de SDS (ver figuras 6(b) y 6(d)). De hecho, la presencia de moléculas de SDS en la región interfacial favorece la migración de las moléculas de alcohol a las fases acuosa y aceite, reduciendo la concentración de estas especies en la región interfacial. Por último, los perfiles de densidad nos permitió determinar que la re- lación SDS/alcohol en la región interfacial fue de 5:4, indicando que una molécula de alcohol ocupa el espacio que existe entre dos moléculas de SDS presentes en la monocapa.
Distribución de las moléculas de 1-butanol y 2-butanol en la región interfacial del sistema agua/SDS/n-octano. Uso de la herramienta gmx-densmap
La configuración final de las moléculas de 1-butanol, 2-butanol y SDS en el plano xy de la región interfacial del sistema agua/n-octano se muestran en la figura 7.
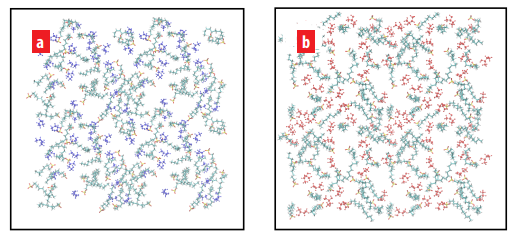
Distribución final del 1-butanol, 2-butanol y SDS en el plano xy localizado en la región interfacial del sistema agua/n-octano. (a) Monocapa formada por SDS con 1-butanol y (b) Monocapa formada por SDS con 2-butanol. Las moléculas de 1-butanol y 2-butanol son mostradas en azul oscuro y rojo, respectivamente.
En ambas figuras 7(a) y 7(b) se puede observar como las moléculas de SDS se distribuyen uniformemente en el plano xy de la región interfacial del sistema agua/n- octano. También, en estas figuras, las moléculas de 1-butanol y 2-butanol se encuentran presentes en la interface. En este caso, estas moléculas se distribuyen en la región interfacial junto con las moléculas de SDS, lo cual demuestra efectivamente que estas moléculas de alcohol coexisten con las moléculas de SDS en la región interfacial.
Para confirmar la distribución de las moléculas de 1-butanol, 2-butanol y SDS, los mapas de densidad en el plano xy de la región interfacial fueron determinados y son mostrados en la figura 8.
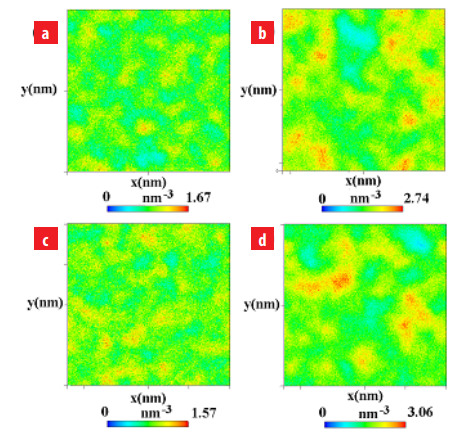
Mapas de la densidad en número del 1-butanol, 2-butanol y SDS en el plano xy de la región interfacial de los sistemas. (a) SDS en agua/SDS+1-butanol/n-octano, (b) 1-butanol en agua/SDS+1-butanol/n-octano, (c) SDS en agua/SDS+2-butanol/n-octano y (d) 2-butanol en agua/SDS+1-butanol/n-octano.
Las figuras 8(a) y 8(b) muestran los mapas de densidad de las moléculas de SDS en el plano xy. Estos mapas de densidad demuestran que el surfactante SDS se distribuye uniformemente en la región interfacial dejando ciertos espacios vacíos en la monocapa. A su vez, las figuras 8(c) y 8(d) muestran los mapas de densidad del 1-butanol y 2-butanol. En este caso, se mues- tra claramente que las moléculas de 1-butanol y 2-butanol ocupan los espacios vacíos entre las moléculas de SDS presentes en la interface. También, se pueden observar algunas zonas de aglo- meramiento de moléculas de alcohol en la región interfacial. Estas zonas de aglomeramientos son más visibles para el 2-butanol en comparación con el 1-butanol. Finalmente, estos mapas de densidad corroboran que las moléculas de alcohol coexisten con las moléculas de SDS en la región interfacial generando una monocapa y película interfacial más estable. En base a es- to, las moléculas de alcohol ubicadas entre las moléculas de SDS permite reducir la repulsión entre los grupos hidrofílicos aumentando la estabilidad de la monocapa localizada en la región interfacial.
CONCLUSIONES
En este trabajo, mediante simulaciones de dinámica molecular, se determinó la distribución de las moléculas de 1-butanol y 2-butanol en los sistemas agua/n-octano y agua/SDS/n-octano usando los perfiles y mapas de densidad.
El perfil de densidad del sistema agua/1-butanol/n-octano demuestra que el 1-butanol se dis- tribuye particularmente en la capa de agua y la región interfacial. En cambio, para el sistema agua/2-butanol/n-octano, el 2-butanol prefiere estar exclusivamente en la región interfacial. A su vez, cuando el SDS esta presente en la interface agua/n-octano, el 1-butanol se distribuye en las capas de agua, n-octano y en la región interfacial. Por el contrario, el 2-butanol esta presente en la capa de n-octano y la región interfacial.
A nivel molecular, los perfiles y mapas de densidad demuestran que las moléculas de 1-butanol, 2-butanol y SDS coexisten en la región interfacial del sistema agua/n-octano. La herramien- ta gmx-density permitió determinar la distribución de las moléculas de 1-butanol y 2-butanol a lo largo del eje z y la herramienta gmx-densmap permitió estimar la distribución de estas moléculas en el plano xy localizado en el centro de la región interfacial.
La presencia de moléculas de 1-butanol y 2-butanol en la monocapa de surfactante incrementa la estabilidad de la película interfacial. En este caso, las moléculas de alcohol ocupan los espacios vacíos entre las moléculas de SDS reduciendo la repulsión entre los grupos hidrofílicos del surfactante.
AGRADECIMIENTOS
Los autores José G. Parra, Elizabeth Perozo y Geraldine Rodriguez agradecen al proyecto Fonacit-Venezuela 2005000424.
CONTRIBUCIONES DE LOS AUTORES
José G. Parra y Yosslen R. Aray coincibieron la idea central dela investigación y desarrollaron la metodología. José G. Parra, Geraldine Rodriguez y Elizabeth Perozo desarrollaron los cálculos en el programa GROMACS. La redacción del manuscrito fue hecha por José G. Parra. También, José G. Parra y Peter Iza participaron en el análisis y discusión de los resultados. Peter Iza y José G. Parra corroboraron los datos obtenidos de las simulaciones.
REFERENCIAS
[1] Myers, D. (2002). Surface activity and surfactant structures. Surfaces, Interfaces, and Co- lloids: Principles and Applications, Second Edition, 21-39.
[2] Rosen, M. J., & Kunjappu, J. T. (2012). Surfactants and interfacial phenomena. John Wiley & Sons.
[3] Myers, D. (2005). Surfactant science and technology. John Wiley & Sons.
[4] Holmberg, K. (2002). Handbook of applied surface and colloid chemistry (Vol. 1). John Wiley & Sons.
[5] Prosser, A. J., & Franses, E. I. (2001). Adsorption and surface tension of ionic surfactants at the air–water interface: review and evaluation of equilibrium models. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 178(1-3), 1-40.
[6] Shah, D. O., (1998). Monolayers: Quarter Century Progress at the University of Florida. Micelles, Microemulsions, and Monolayers, 1.
[7] Ulman, A. (2013). An Introduction to Ultrathin Organic Films: From Langmuir-Blodgett to Self-Assembly. Academic press.
[8] Petersen, K. S., & Christensen, P. L. (2007). Phase Behavior of Petroleum Reservoir Fluids. CRC Press, Boca Raton, London and New York.
[9] Maag, H. (1984). Fatty acid derivatives: important surfactants for household, cosmetic and industrial purposes. Journal of the American Oil Chemists Society, 61(2), 259-267.
[10] Ikeda, S., Ozeki, S., & Hayashi, S. (1980). Size and shape of charged micelles of ionic surfactants in aqueous salt solutions. Biophysical Chemistry, 11(3-4), 417-423.
[11] Hayter, J. B., & Penfold, J. (1981). Self-consistent structural and dynamic study of con- centrated micelle solutions. Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases, 77(8), 1851-1863.
[12] Fernandez, P., Schrodle, S., Buchner, R., & Kunz, W. (2003). Micelle and solvent relaxation in aqueous sodium dodecylsulfate solutions. ChemPhysChem, 4(10), 1065-1072.
[13] Valkovska, D. S., Shearman, G. C., Bain, C. D., Darton, R. C., & Eastoe, J. (2004). Adsor- ption of ionic surfactants at an expanding air-water interface. Langmuir, 20(11), 4436-4445.
[14] Taylor, D. J. F., Thomas, R. K., & Penfold, J. (2002). The adsorption of oppositely charged polyelectrolyte/surfactant mixtures: neutron reflection from dodecyl trimethylammonium bro- mide and sodium poly (styrene sulfonate) at the air/water interface. Langmuir, 18(12), 4748- 4757.
[15] Li, Z. X., Dong, C. C., & Thomas, R. K. (1999). Neutron reflectivity studies of the surface excess of gemini surfactants at the air-water interface. Langmuir, 15(13), 4392-4396.
[16] Reiss-Husson, F., & Luzzati, V. (1964). The structure of the micellar solutions of some amphiphilic compounds in pure water as determined by absolute small-angle X-ray scattering techniques. The Journal of Physical Chemistry, 68(12), 3504-3511.
[17] Rohde, A., & Sackmann, E. (1979). Quasielastic light-scattering studies of micellar so- dium dodecyl sulfate solutions at the low concentration limit. Journal of Colloid and Interface Science, 70(3), 494-505.
[18] Jusufi, A., LeBard, D. N., Levine, B. G., & Klein, M. L. (2012). Surfactant concentration effects on micellar properties.The Journal of Physical Chemistry B, 116(3), 987-991.
[19] Vollhardt, D., & Emrich, G. (2000). Coadsorption of sodium dodecyl sulfate and medium- chain alcohols at the air- water interface. Colloids and Surfaces A: Physicochemical and Engi- neering Aspects, 161(1), 173-182.
[20] Srinivasan, V., & Blankschtein, D. (2003). Effect of Counterion Binding on Micellar So- lution Behavior: 1. Molecular- Thermodynamic Theory of Micellization of Ionic Surfactants. Langmuir, 19(23), 9932-9945.
[21] Zhang, S., Zhu, P., Sun, Y., Yang, Y., Cao, X., Song, X., & Li, Y. (2014). Study of the mo- lecular array behaviour of laurel alkanolamide at the oil–water interface and the high interfacial activity enhanced by an inherent synergistic effect. RSC Advances, 4(79), 41831-41837.
[22] Chai, J. L., Wu, Y. T., Li, X. Q., Yang, B., & Lu, J. J. (2011). Effect of oil/water ratios on the phase behavior and the solubilization ability of microemulsion systems containing sodium dodecyl sulfate. Journal of Solution Chemistry, 40(11), 1889-1898.
[23] Gunaseelan, K., Umlong, I. M., Mukhim, T., & Ismail, K. (2003). Electrical conductance behavior of oil-in-water microemulsions stabilized by sodium dodecyl sulfate and 1-butanol. Langmuir, 19(18), 7276-7281.
[24] Salager, J. L., Antón, R. E., Sabatini, D. A., Harwell, J. H., Acosta, E. J., & Tolosa, L. I. (2005). Enhancing solubilization in microemulsions-state of the art and current trends. Journal of Surfactants and Detergents, 8(1), 3-21.
[25] Salager, J. L., Forgiarini, A. M., & Bullón, J. (2013). How to attain ultralow interfacial tension and three-phase behavior with surfactant formulation for enhanced oil recovery: a re- view. Part 1. Optimum formulation for simple surfactant- oil-water ternary systems. Journal of Surfactants and Detergents, 16(4), 449-472.
[26] Baviere, M., Schechter, R., & Wade, W. (1981). The influence of alcohols on microemul- sion composition. Journal of Colloid and Interface Science, 81(1), 266-279
[27] Kahlweit, M., Strey, R., & Busse, G. (1991). Effect of alcohols on the phase behavior of microemulsions. The Journal of Physical Chemistry, 95(13), 5344-5352.
[28] Jones, S. C., & Dreher, K. D. (1976). Cosurfactants in micellar systems used for tertiary oil recovery. Society of Petroleum Engineers Journal, 16(03), 161-167.
[29] Jang, S. S., Lin, S. T., Maiti, P. K., Blanco, M., Goddard,W. A., Shuler, P., &Tang,Y. (2004). Molecular dynamics study of a surfactant-mediated decane-water interface: effect of molecular architecture of alkyl benzene sulfonate. The Journal of Physical Chemistry B, 108(32), 12130-12140.
[30] Chanda, J., & Bandyopadhyay, S. (2006). Molecular dynamics study of surfactant mono- layers adsorbed at the oil/ water and air/water interfaces. The Journal of Physical Chemistry B, 110(46), 23482-23488.
[31] Wu, R., Deng, M., Kong, B., Wang, Y., & Yang, X. (2009). Molecular dynamics simulations of ammonium surfactant monolayers at the heptane/water interface. The Journal of Physical Chemistry B, 113(38), 12680-12686.
[32] Qu, G., Xue, C., Zhang, M., Liang, S., Han, Y., & Ding, W. (2016). Molecular dynamics simulation of sulfobetaine-type zwitterionic surfactants at the decane/water interface: structure, interfacial properties. Journal of Dispersion Science and Technology, 37(12), 1710-1717.
[33] Mills, A. J., Wilkie, J., & Britton, M. M. (2014). NMR and molecular dynamics study of the size, shape, and composition of reverse micelles in a cetyltrimethylammonium bro- mide (CTAB)/n-hexane/pentanol/water microemulsion. The Journal of Physical Chemistry B, 118(36), 10767-10775.
[34] Parra, J., & Aray, Y. (2016). Comportamiento del SDS localizado en la región interfacial agua/n-octano. Un estudio usando dinámica molecular, ACI Avances en Ciencias e Ingenierías, 8(14), 98-110.
[35] Wang, L., Hu, Y., Liu, R., Liu, J., & Sun, W. (2017). Synergistic adsorption of DDA/alcohol mixtures at the air/water interface: A molecular dynamics simulation. Journal of Molecular Liquids, 243, 1-8.
[36] Liu, Z. Y., Xu, Z., Zhou, H., Wang, Y., Liao, Q., Zhang, L., & Zhao, S. (2017). Interfa- cial behaviors of betaine and binary betaine/carboxylic acid mixtures in molecular dynamics simulation. Journal of Molecular Liquids, 240, 412-419.
[37] Domínguez, H. (2006). Computer studies on the effects of long chain alcohols on sodium dodecyl sulfate (SDS) molecules in SDS/dodecanol and SDS/hexadecanol monolayers at the air/water interface. The Journal of Physical Chemistry B, 110(26), 13151-13157.
[38] Méndez-Bermúdez, J. G., & Dominguez, H. (2016). Structural changes of a sodium do- decyl sulfate (SDS) micelle induced by alcohol molecules. Journal of Molecular Modeling, 22(1), 33.
[39] Oostenbrink, C., Villa, A., Mark, A. E., & Van Gunsteren, W. F. (2004). A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force field parameter sets 53A5 and 53A6. Journal of Computational Chemistry, 25(13), 1656-1676.
[40] Berendsen, H. J., Postma, J. P., van Gunsteren, W. F., & Hermans, J. (1981). Interaction models for water in relation to protein hydration. In Intermolecular forces (pp. 331-342). Sprin- ger, Dordrecht.
[41] Frenkel, D.; & Smith, B. (1996). Understanding Molecular Simulation: From Algorithms to Applications. Academic Press, Inc: San Diego.
[42] Malde, A. K., Zuo, L., Breeze, M., Stroet, M., Poger, D., Nair, P. C., & Mark, A. E. (2011). An automated force field topology builder (ATB) and repository: version 1.0. Journal of Che- mical Theory and Computation, 7(12), 4026-4037.
[43] Kirkwood, J. G., & Buff, F. P. (1949). The statistical mechanical theory of surface tension. The Journal of Chemical Physics, 17(3), 338-343.
[44] Neyt, J. C., Wender, A., Lachet, V., Ghoufi, A., & Malfreyt, P. (2014). Quantitative Pre- dictions of the Interfacial Tensions of Liquid–Liquid Interfaces through Atomistic and Coarse Grained Models. Journal of Chemical Theory and Computation, 10(5), 1887-1899.
[45] Hanwell, M. D., Curtis, D. E., Lonie, D. C., Vandermeersch, T., Zurek, E., & Hutchison, G. R. (2012). Avogadro: an advanced semantic chemical editor, visualization, and analysis plat- form. Journal of Cheminformatics, 4(1), 17.
[46] Hess, B., Kutzner, C., Van Der Spoel, D., & Lindahl, E. (2008). GROMACS 4: algo- rithms for highly efficient, load- balanced, and scalable molecular simulation. Journal of Che- mical Theory and Computation, 4(3), 435-447.
[47] Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., & Berendsen, H. J. (2005). GROMACS: fast, flexible, and free. Journal of Computational Chemistry, 26(16), 1701-1718.
[48] Lindahl, E., Hess, B., & Van Der Spoel, D. (2001). GROMACS 3.0: a package for mole- cular simulation and trajectory analysis. Molecular Modeling Annual, 7(8), 306-317.
[49] Bussi, G., Donadio, D., & Parrinello, M. (2007). Canonical sampling through velocity rescaling. The journal of Chemical Physics, 126(1), 014101.
[50] Berendsen, H. J., Postma, J. V., van Gunsteren, W. F., DiNola, A. R. H. J., & Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics, 81(8), 3684-3690.
[51] Hess, B., Bekker, H., Berendsen, H. J., & Fraaije, J. G. (1997). LINCS: a linear constraint solver for molecular simulations. Journal of Computational Chemistry, 18(12), 1463-1472.
[52] Hockney, R. W., Goel, S. P., & Eastwood, J. W. (1974). Quiet high-resolution computer models of a plasma. Journal of Computational Physics, 14(2), 148-158.
[53] Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., & Pedersen, L. G. (1995). A smooth particle mesh Ewald method. The Journal of Chemical Physics, 103(19), 8577-8593.
[54] http://plasma-gate.weizmann.ac.il/grace/.
[55] Humphrey, W., Dalke, A., & Schulten, K. (1996). VMD: visual molecular dynamics. Jour- nal of Molecular Graphics, 14(1), 33-38.
[56] Cordeiro, N. (2003). Interfacial Tension Behaviour of Water/Hydrocarbon Liquid-Liquid Interfaces: A Molecular Dynamics Simulation. Molecular Simulation, 29(12), 817-827.
[57] Aray,Y., Parra, J. G., Jiménez, D. M., Paredes, R., Martiz, A., Samaniego, S., Cornejo, M., Ludena, E. & Paredes, C. (2017). Exploring the effect of the O-(1-heptylnonyl) benzene sulfonate surfactant on the nature of the linear hydrocarbons/ water interface by means of an atomistic molecular dynamics simulation. Journal of Computational Methods in Sciences and Engineering, 17(1), 39-53.
[58] Aveyard, R., & Haydon, D. A. (1973). An introduction to the principles of surface che- mistry. CUP Archive.
[59] Goebel, A., & Lunkenheimer, K. (1997). Interfacial tension of the water/n-alkane interface. Langmuir, 13(2), 369-372.
[60] Lu, J. R., Marrocco, A., Su, T. J., Thomas, R. K., & Penfold, J. (1993). Adsorption of dodecyl sulfate surfactants with monovalent metal counterions at the air-water interface studied by neutron reflection and surface tension. Journal of Colloid and Interface Science, 158(2), 303-316.
[61] Rehfeld, S. J. (1967). Adsorption of sodium dodecyl sulfate at various hydrocarbon-water interfaces. The Journal of Physical Chemistry, 71(3), 738-745.